

Clinical Medical
Reviews and Case Reports
General and Oral Aspects in Apert Syndrome: Report of a Case
Villarreal-Becerra Einer1,2*, Guerrero- Guevara Rember1, Espías-Gómez Angel2, Leonard RH Jr3 and Chimenos-Küstner Eduardo4
1Department of Dentistry, Antenor Orrego Private University, Peru
2Department of Dentistry, University of Barcelona, Spain
3Department of Diagnostic Sciences and General Dentistry, University of North Carolina, USA
4Department of Oral Medicine, University of Barcelona, Spain
*Corresponding author: Villarreal-Becerra Einer, L'Hospitalet de Llobregat, Bellvitge, Calle Feixa Llarga, s/n, 08907, Pavelló de Govern, Spain, E-mail: evillarrealb@upao.edu.pe
Clin Med Rev Case Rep, CMRCR-2-050, (Volume 2, Issue 8), Case Report; ISSN: 2378-3656
Received: March 12, 2015 | Accepted: August 24, 2015 | Published: August 26, 2015
Citation:Villarreal-Becerra E, Sánchez-Soler L, Espías-Gómez A, Leonard RH Jr, Herrero-Payo J, Eduardo CK (2015) General and Oral Aspects in Apert Syndrome: Report of a Case.. Clin Med Rev Case Rep 2:050. 10.23937/2378-3656/1410050
Copyright: © 2015 Villarreal-Becerra E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Background: The present paper describes the general and oral manifestations in a 32-year-old man previously diagnosed with Apert syndrome.
Clinical examination revealed features of acrocephalosyndactyly. The patient was found to have a flattened occiput with frontal prominence, abnormal contour of head (brachycephaly), shallow and downward slanting orbits with bilateral proptosis, hypertelorism, retruded midface, and prognathic mandible.Dental anormalies were present in a patient. Intraoral evaluation revealed normal mouth opening with anterior severe skeletal open bite and Byzantine-arch palate, maxillary alveolar ridges with crowding of maxillary and mandibular teeth, poor hygiene with heavy dental calculus and periodontal pseudopocket, dental caries, severe anterior open bite and crossbite, macroglosia and smooth tongue.
The high prevalence of dental anomalies and ectopic eruption may suggest a possible etiologic relationship with the Apert syndrome.
Keywords
Acrocephalosyndactylia, Craniosynostosis, Tooth abnormalities, Mouth abnormalities, Apert Syndrome
Introduction
Apert syndrome is a rare congenital type I acrocephalosyndanctyly syndrome, characterized by craniosynostosis (premature fusion of cranial sutures), severe syndactyly of the hands and feet, symphalangism and dysmorphic facial features [1]. Premature fusion of cranial sutures restricts growing in the region of fused sutures and leads to craniofacial abnormalities, including calvarial shape. Various extracranial manifestations are present. The prevalence of this disease is 15.5 per million live births and accounts for 4.5% of all cases of craniosynostosis [2]. Craniosynsostosis syndromes exhibit considerable phenotypic and genetic heterogeneity. Sagittal synostosis is common form of isolated craniosynostosis. The sutures involved, the shape of the skull and associated malformations give a clue to the specific diagnosis. Apert syndrome is one of the most serious of these syndromes. Most syndromic craniosynostosis require multidisciplinary Management [3].
Apert syndrome (acrocephalosyndactylia) is a developmental malformation characterized by craniosynostosis, a cone-shaped calvarium (acrocephaly), hypertelorism, midface hypoplasia, pseudo cleft-palate, a parrot beak shaped nose, pharyngeal attenuation, and syndactyly of the hands and feet [3,4]. The inheritance of Apert's syndrome is autosomal dominant with the locus of a mutation of FGFR2 on chromosome 10q (10q25- 26). Suture progenitor cells with fibroblast growth factor receptors (FGFR2) that have undergone a mutation cannot transduce signals from extracellular fibroblast growth factors (FGFs). Therefore, these cells do not receive the signal to produce the necessary fibrous material essential for a normal calvarial suture [5].
Apert syndrome was first reported by Wheaton in 1894 and French pediatrician Eugene Apert published a series of nine cases in 1906 [3,6]. Most cases are sporadic, with an incidence of 1:160 000; however due to high infant mortality, the incidence in the general population is lower. Advanced male parental age has been consistently noted [7]. During the course of the disease, growth and mental retardation can be observed [4,7].
In Apert cases, the spheno-occipital and spheno-ethmoidal synchondroses and the fronto-ethmoidal suture fuse early, resulting in a severely shortened posterior cranial base and a relatively short anterior cranial base with a resultant hypoplastic midface. Consistent with the observation of midface hypoplasia, the maxilla also exhibits a transverse deficiency [5]. The most readily observed malocclusions are a severe maxillary anterior open bite and a severely crowded and retrusive maxillary arch due to the constricted secondary palate [5].
The maxillary alveolar arch is V-shaped [8]. Due to the narrower maxillary arch, bilateral or unilateral posterior crossbite can be observed [9-11]. Cleft of the soft palate is observed in 30% of the cases [12,13]. In mouse models exist data that reveal significantly abnormal cell patterns of proliferation, differentiation and apoptosis local to the inter-premaxillary suture, causing a lack of fusion and maldevelopment of the anterior palate in Fgfr2+/S252W mice [9,14-18].
The nose is short and broad with a bulbous tip and nasolabial angle is diminished [19]. The midface hypoplasia contributes to retruded middle third of the face, resulting in relative mandibular prognathism [15,16,19]. The lips frequently assume a trapezoid configuration because the upper lip is lifted in the midline [20,21].
Impactions, severe crowding of developing teeth within the alveolus, delayed eruption, thick gingiva, and sometimes supernumerary teeth or congenitally missing teeth are the hall marks of maxillary dental development in Apert patients [22]. The nasopharyngeal and oropharyngeal attenuation cause Apert's individuals to become mouth breathers with a resultant anterior open bite [22].
The familial pattern, equal number of affected males and females, and increased paternal age in sporadic cases strongly suggest autosomal dominant inheritance [23,24].
Treatment involves multidisciplinary teamwork including craniofacial surgeon, neurosurgeon, neurologist, ENT (ear, nose, and throat), audiologist, pediatrician, speech pathologist, oral surgeon, psychologist, periodontist and an orthodontist [25,26]. Surgical care involves early release of the coronal suture and frontoorbital advancement with reshaping to allow proper brain growth and reduce dysmorphic and unwanted skull growth changes [25]. Craniotomy is often performed during the 1st year of life to treat the craniosynostosis. Frontofacial advancement and midface advancement can be performed later to correct the proptosis and midface hypoplasia. Coordinated orthodontic therapy is often necessary to bring unerupted teeth into place and improve occlusion [27,28].
Case Report
A 32- year- old male presented with the complaints of malaligned teeth, heavy dental calculus, difficulty in chewing food, and craniofacial and limbs deformity.
He was the first child born to nonconsanguineous parents. Pregnancy and labor were uneventful and there was no history of taking any drugs during the entire term of pregnancy. His mother was 45 years old and his father was 52 years old. The family history contained no report of similar cases.
At birth, the child had craniosynostosis, brachycephaly, and syndactyly of hands and feet.
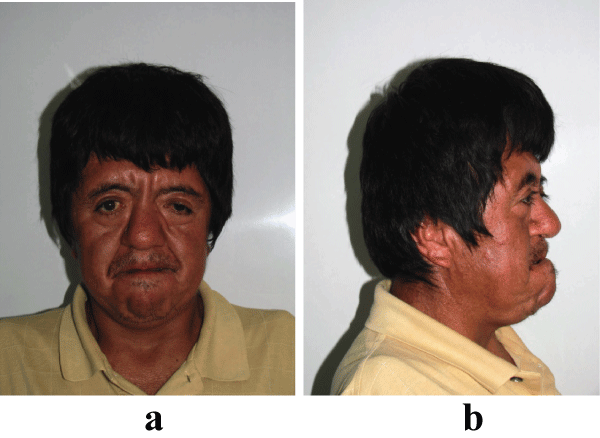
Clinical examination revelaed features of acrocephalosyndactyly. The patient was found to have a flattened occiput with frontal prominence, abnormal contour of head (brachycephaly), shallow and downward slanting orbits with bilateral proptosis, hypertelorism, retruded midface, and prognathic mandible (Figures1a and Figure1b).
.
Figure 1: (a) Extraoral front view of patient showing hypertelorism, parrot beak nose, depressed nasal bridge, and downward alanting outer canthus of eyes. (b) Maxillary hypoplasia results in psudoprognatism.
View Figure 1
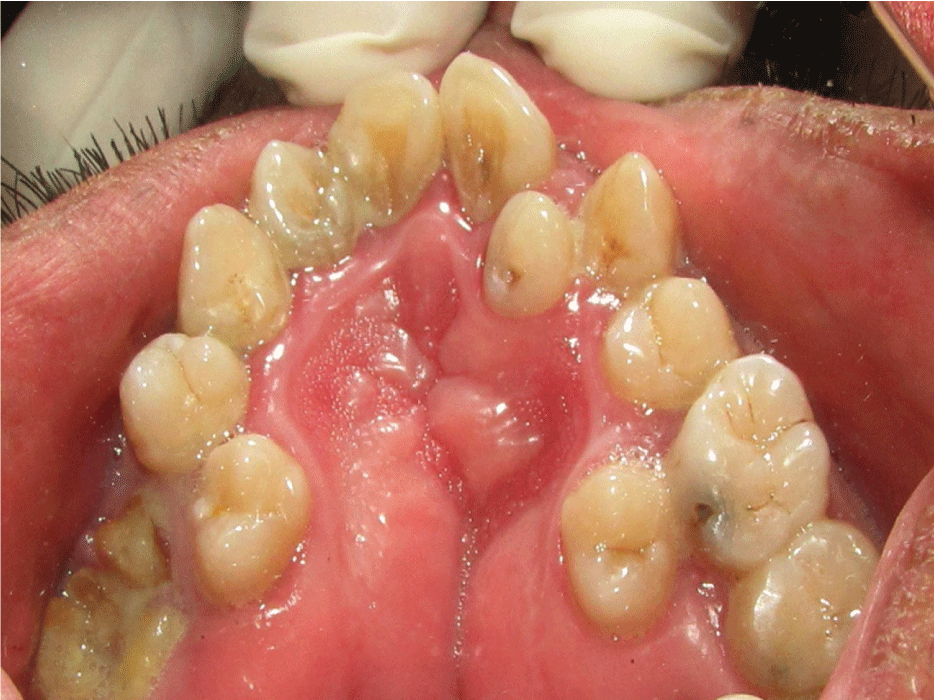
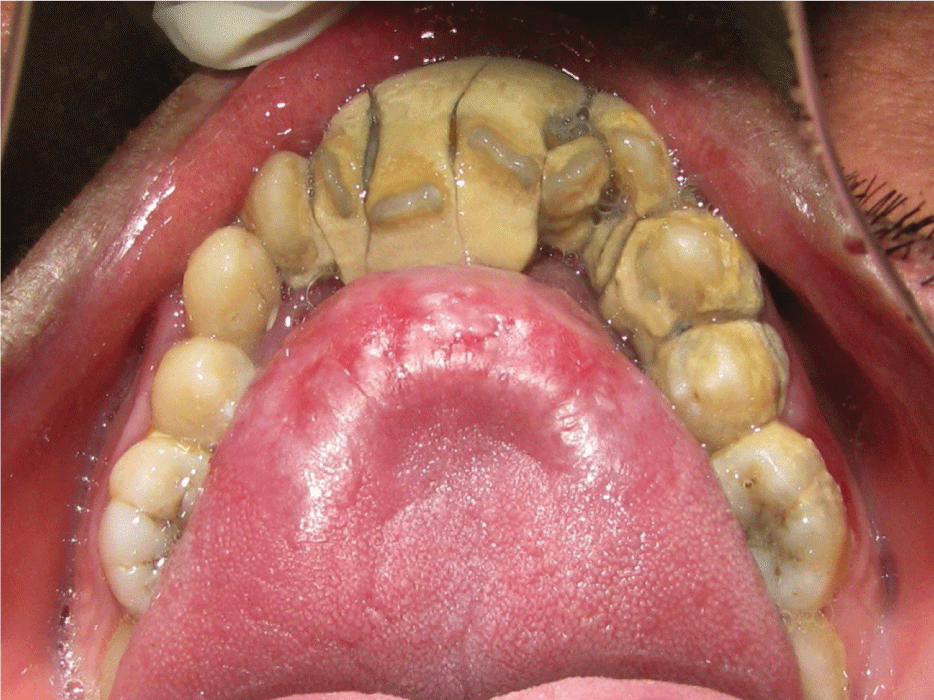
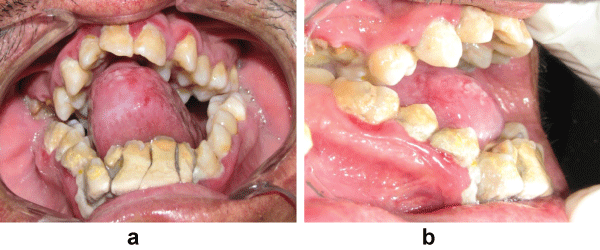
Intraoral examination showed normal mouth opening with anterior severe skeletal open bite and a high arched (V-shaped) palatal vault or Byzantine-arch palate associated with lateral swellings of the palatine processes, one on either side of the middle miming a pseudocleft in the midline (Figure 2). Maxillary alveolar ridges were thick with crowding of maxillary and mandibular teeth. Heavy dental calculus, congestion and swelling of the gingiva and periodontal pseudopockets associated with anterior and posterior teeth. Dental caries on anterior and posterior teeth were present. Severe maxillary and mandibular dental crowding, with the rotation of maxillary central incisors and the palatal position of second bicuspids and lateral incisors were observed. At the occlusal examination, severe anterior open bite and crossbite were observed, only the first and second molars being involved in centric occlusion and macroglosia and smooth tongue were observed too (Figures 2, Figure 3 and Figure 4).
.
Figure 2: Intraoral maxillary view showing Byzantine-arch palate, pseudocleft in the midline, crowding of teeth, caries and root fragment.
View Figure 2
.
Figure 3: Intraoral maxillary view showing crowding of teeth, macroglosia and heavy dental calculus.
View Figure 3
On investigation, panoramic view radiograph showed deformity of jaws with severe skeletal open bite, partially erupted lower right wisdom molar , and presence and root fragments of first and second upper right molars (Figure 5).
.
Figures 4 (a and b): Intraoral front and lateral views showing swelling of gingiva, crowding of teeth, macroglosia heavy dental calculus. severe anterior open bite and crossbite.
View Figure 4
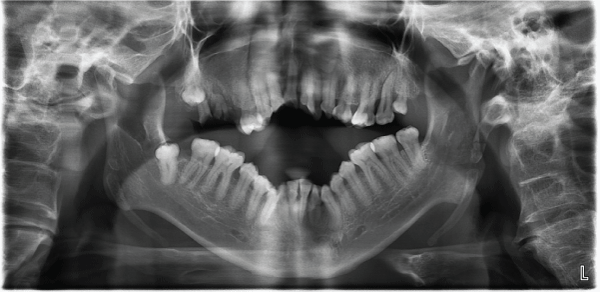
.
Figure 5: Panoramic view showing non- erupted third molar, open skeletal bite and root frgaments.
View Figure 5
He had symmetrical syndactyly with complete fusion of all digits of hands (except thumb) and feet (Figure 6). The systemic examination revealed that patient has some ophthalmologic, ear, and central nervous system abnormalities and mild degree of mental deficiency have been observed.
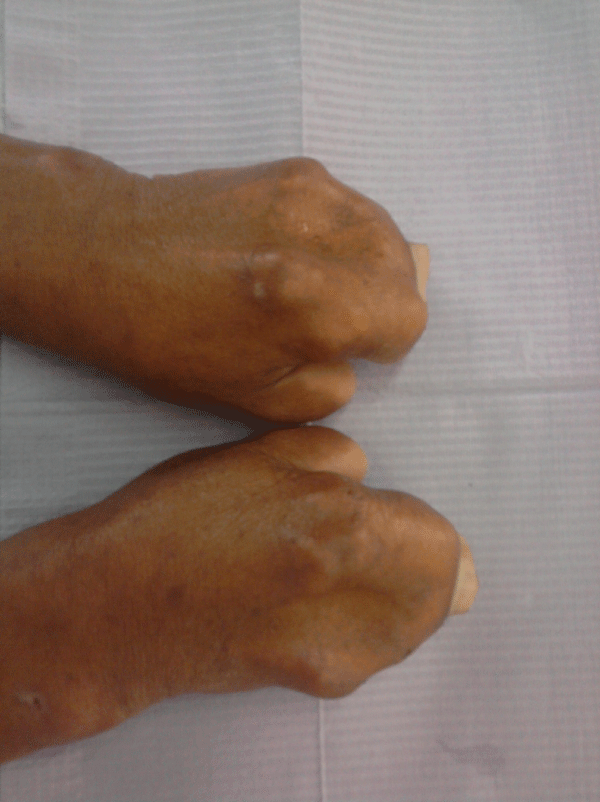
Discussion
This patient demonstrated the clinical triad that characterizes Apert syndrome: Brachycephalic skull, midface hypoplasia and syndactyly of hands and feet [12].
Craniosynostosys refers to a premature fusion of the calvarial sutures. Historically, the clinical description of craniosynostosys date back to Hippocrates and Galen, but first historical reference to craniosynostosys was made by Mestrius Plutarchus (46-127 AD) [29]. The identification of two pre-Columbian skulls with sagittal synostosys (dated at 6000 and 250 BC) confirms that craniosynostosys is an ancient disorders of humans [30]. Wheaton SW, in 1894, described the first two cases of Apert syndrome revealing craniofacial, skull base and limbs findings, but unfortunately he attributed the calvarial phenotype and respiratory deficiencies to con- genital syphilis and the syndactyly to fetal inflammation [31]. Twelve years later, the Dr. Eugene Charles Apert described nine cases of syndactyly associated with acrocephaly [32].
The clinical features of Apert syndrome are distinctive. The coronal suture fuses prematurely, at least three month leading, as we described earlier, to an acrocephalic (cone-shaped) head with a flattened occiput, shortened anterior-posterior diameter, a high prominent forehead, a characteristic form of the nose and of the mouth. The midface of these patients is hypoplastic. Occular anomalies as hypertelorism, proptosis, strabismus and down slanting palpebral fissures are often present and are due to shortening of the bony orbit [33,34]. Our patient presented some of the above mentioned features [1]. Commonly associated systemic features include cardiac anomalies, visual and hearing defects [13]. This is in accordance with the present case.
A pseudocleft due to the accumulation of the proliferated lateral palatal tissue mass was recorded for our patient. In Apert syndrome cases, the swellings are usually present in infancy and increase in mass as the child growth older. The cumulative tissues can proliferate to such an extent as to lead sometimes to a mis- taken diagnosis of cleft palate. The prevalence of a real cleft palate was reported between 25 to 75% of Apert subjects [34].
Failure in the anteroposterior and downward growth of the maxilla causes the maxillary hypoplasia and a resultant contraction of nasopharyngeal airway [22].
In patients with Apert syndrome, severe skeletal Class III open bite malocclusion can be observed due to the maxillary deficiency and the inclination of the upper jaw. Therefore, it is usually necessary to add ortognathic surgery to the treatment plan. The infant is likely to suffer from oral hygiene problems during treatment [22].
The maxillary dental arch is v-shaped and there can be some compensatory growth of the alveolar base. Most probably, the alveolus thickens to accommodate the teeth that are impacted and crowded to an extreme degree in a small maxilla. The maxilla slants down posteriorly. As a result, open bite is common, if untreated, the maxilla-mandibular discrepancy and class III malocclusion worsens with age [35].
The appearance of a patient with Apert syndrome is prognathic. The "pseudoprognathic" appearance is basically due to maxillary retroposition. Impactions, severe crowding of developing teeth within the alveolus, delayed eruption, thick gingival and sometimes supernumerary teeth or congenitally missing teeth are the hallmarks of the maxillary dental development in the Apert patient. There is severe arch length deficiency to accommodate the tooth material. There is a mean dental developmental delay of 0.96 years, with a range of 0.5 to 2.9 years. It is postulated that mutation in the FGFR2 gene has an effect on the mesenchymal development, which has an effect on tooth morphogenesis [35].
For the patient with Apert syndrome, oral hygiene is as important as it is difficult. Hand deformities make it difficult to brush the teeth. The new generation of electric tooth brushes and fluoride mouth rinses may make the task easier. Professional care including frequent dental examination, oral hygiene prophylaxis, fluoride treat- ments, and dental sealants are very important [6].
The fact that the therapeutic management of patients with Apert syndrome should be multidisciplinary is a logical consequence of the previously exposed facts. These patients generally require lifelong management by a multidisciplinary team of health care specialists [1].
Conclusions
The rarity of the AS, the typical craniofacial and dental features, remains a major medical condition with considerable morbidity. In the treatment plan, the odontological aspect plays an important role for the management of preventable oral diseases such as dental caries and periodontal disease.
The information and the motivation of the parents regarding the necessity of the treatment and the extensive use of home prevention methods are essential.This case report attempts to throw some light on this rare syndrome.
Competing Interests
The authors declare that they do not have any competing or financial interests.
References
-
DeGiovanni CV, Jong C, Woollons A (2007) What syndrome is this? Apert syndrome. Pediatr Dermatol 24: 186-188.
-
Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, Mastroiacovo P, et al. (1992) Birth prevalence study of the Apert syndrome. Am J Med Genet 42: 655-659.
-
Gleeson JG, Dobyns WB, Plawner L, Ashwal S (2006) Congenital structural defects. In: Swaiman KF, Ashwal S, Ferriero DM Pediatric Neurology: Principles and Practice. (4th edn), Philadelphia: Mosby Elsevier 441-454.
-
OMIM (TM), Bethesda (2008) McKusick-Nathans Institute for Genetic Medicine, JohnHopkins University, National Center for Biotechnology Information, National Library of Medicine, Online Mendelian Inheritance in Man.
-
Wilkie AO, Bochukova EG, Hansen RM, Taylor IB, Rannan Eliya SV, et al. (2007) Clinical dividends from the molecular genetic diagnosis of craniosynostosis. Am J Med Genet A 143A: 1941-1949.
-
Bonaventure J, E Ghouzzi V (2003) Molecular and cellular bases of syndromic craniosynostoses. Expert Rev Mol Med 5: 1-17.
-
Kapp-Simon KA, Speltz ML, Cunningham ML, Patel PK, Tomita T (2007) Neurodevelopment of children with single suture craniosynostosis: a review. Childs Nerv Syst 23: 269-281.
-
Jadico SK, Huebner A, McDonald-McGinn DM, Zackai EH, Young TL (2006) Ocular phenotype correlations in patients with TWIST versus FGFR3 genetic mutations. J AAPOS 10: 435-444.
-
Cohen MM Jr, Kreiborg S (1996) A clinical study of the craniofacial features in Apert syndrome. Int J Oral Maxillofac Surg 25: 45-53.
-
Chen L, Li D, Li C, Engel A, Deng CX (2003) A Ser252Trp [corrected] substitution in mouse fibroblast growth factor receptor 2 (Fgfr2) results in craniosynostosis. Bone 33: 169-178.
-
Reardon W (2000) Craniosynostosis. Diagnosis, evaluation and management J Med Genet 37: 727.
-
Paravatty RP, Ahsan A, Sebastian BT, Pai KM, Dayal PK (1999) Apert syndrome: a case report with discussion of craniofacial features. Quintessence Int 30: 423-426.
-
Amar T, Krishna V, Sona K (2007) Apert syndrome: A rare presentation. J Indian Acad Clin Med 8: 245-246.
-
Fanganiello RD, Sertié AL, Reis EM, Yeh E, Oliveira NA, et al. (2007) Apert p.Ser252Trp mutation in FGFR2 alters osteogenic potential and gene expression of cranial periosteal cells. Mol Med 13: 422-442.
-
Carinci F, Pezzetti F, Locci P, Becchetti E, Carls F, et al. (2005) Apert and Crouzon syndromes: clinical findings, genes and extracellular matrix. J Craniofac Surg 16: 361-368.
-
Pereira V, Sacher P, Ryan M, Hayward R (2009) Dysphagia and nutrition problems in infants with apert syndrome. Cleft Palate Craniofac J 46: 285-291.
-
Upadhyaya V, Upadhyaya DN, Sarkar S (2005) Apert's syndrome: A case report. Indian J Radiol Imaging 15: 477-480.
-
Alp E, Alp H, Koc H, Ucar C, Cimen D (2007) Apert syndrome. Tür- kiye Klinikleri J Pediatr 16: 264-268.
-
Martelli H Jr, Paranaíba LM, de Miranda RT, Orsi J Jr, Coletta RD (2008) Apert syndrome: report of a case with emphasis on craniofacial and genetic features. Pediatr Dent 30: 464-468.
-
Hoover GH, Flatt AE, Weiss MW (1970) The hand and Apert's syndrome. J Bone Joint Surg Am 52: 878-895.
-
Rynearson RD (2000) Case report: orthodontic and dentofacial orthopedic considerations in Apert's syndrome. Angle Orthod 70: 247-252.
-
Ferraro NF (1991) Dental, orthodontic, and oral/maxillofacial evaluation and treatment in Apert syndrome. Clin Plast Surg 18: 291-307.
-
Sannomiya EK, Reis SA, Asaumi J, Silva JV, Barbara AS, et al. (2006) Clinical and radiographic presentation and preparation of the prototyping model for pre-surgical planning in Apert's syndrome. Dentomaxillofac Radiol 35: 119-124.
-
Breugem CC, Fitzpatrick DF, Verchere C (2008) Monozygotic twins with Apert syndrome. Cleft Palate Craniofac J 45: 101-104.
-
Spruijt B, Rijken BFM, Bredero-Boelhouwer HH, Pullens B, Lequin MH, et al. (2015) Atypical presentation of a newborn with Apert syndrome. Child's Nervous System 31: 481-486.
-
Batra P, Duggal R, Parkash H (2002) Dentofacial characteristics in Apert syndrome: a case report. J Indian Soc Pedod Prev Dent 20: 118-123.
-
Madhura D, Naresh S (2010) Apert's syndrome: A rare case report. J Indian Acad Oral Med Radiol 22: 232-235.
-
Cunningham ML, Seto ML, Ratisoontorn C, Heike CL, Hing AV (2007) Syndromic craniosynostosis: from history to hydrogen bonds. Orthod Craniofac Res 10: 67-81.
-
Gerszten PC, Gerszten E, Allison MJ (1998) Diseases of the skull in pre-Columbian South American mummies. Neurosurgery 42: 1145-1151.
-
Wheaton SW (1894) Two specimens of congenital cranial deformity in infants associated with fusion of the fingers and toes. Trans Pathol Soc London 45: 238-241.
-
APERT ME (1906) De l'acrocephalosyndactyly. Bull Soc Méd Hôp Paris 23: 1310.
-
Freiman A, Tessler O, Barankin B (2006) Apert syndrome. Int J Dermatol 45: 1341-1343.
-
Necula V, Cosgarea M, Pop A, Blaga L (2007) Sindromul Apert - prezentare de caz. Revista Româna de Chirurgie Rino- sinusala 46-50.
-
Athanasiadis AP, Zafrakas M, Polychronou P, Florentin-Arar L, Papasozomenou P, et al. (2008) Apert syndrome: the current role of prenatal ultrasound and genetic analysis in diagnosis and counselling. Fetal Diagn Ther 24: 495-498.
-
Premalatha, Kannan VP, Madhu (2010) Apert syndrome. J Indian Soc Pedod Prev Dent 28: 322-325.
