Immune-mediated necrotizing myopathy (IMNM) is a relatively new disease, described for the first time in 2004. IMNM is a form of myositis since it is an inflammatory myopathy with strict involvement of the skeletal muscles albeit lymphocytic infiltration is sparse or absent. The clinical picture, pathological features and autoantibodies (Abs) of IMNM differ markedly from those of antisynthetase syndrome and non-specific myositis. Anti-HMGCR and anti-SRP Abs are strongly specific for IMNM, but their role in the onset and/or persistence of disease is unknown. IMNM is characterized by rapidly progressive proximal muscle weakness, markedly elevated CK levels, and poor responsiveness to corticosteroid therapy.
We present a clinical case of patient with immune-mediated necrotizing myopathy with positive anti-SRP autoantibodies and typical clinical presentation.
Autoimmune myopathy, Necrotizing myositis, Anti-SRP antibodies
Idiopathic inflammatory myopathies (IIMs) are a small group of acquired myopathies that are associated with muscle weakness, increased serum creatine kinase (CK) and myopathic findings on electromyography [1,2]. Immune-mediated necrotizing myopathy (IMNM) or necrotizing autoimmune myopathy (NAM) represents a heterogeneous group of diseases that are characterized by the presence of necrotic fibers on muscle biopsy along with elevated serum CK levels and an abnormal electromyogram (EMG) result [1,2]. Anti-signal recognition particle (anti-SRP) and anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase (anti-HMGCR) antibodies are the most common antibodies associated with NAM [2,3].
Anti-signal recognition particle (anti-SRP) autoantibodies are myositis-specific antibodies found in 4-6% of patients with IIMs [4,5]. These antibodies are directed against SRP, a ribonuclear protein particle that regulates protein translocation across the endoplasmic reticulum membrane during protein synthesis. Studies have demonstrated that anti-SRP myopathy appears distinct form polymyositis and other IIMs by its clinical features and histopathology [6-8]. Patients with anti-SRP antibodies often present clinically with a severe myopathy characterized by markedly elevated serum creatine kinase levels and rapidly progressive proximal muscle weakness leading to significant disability. On histopathology, anti-SRP patients demonstrate a necrotizing myopathy without primary inflammation [6-8]. Anti-SRP myopathy also differs from other immune-mediated myopathies by its characteristically poor response to steroid monotherapy and conventional immunosuppressive therapies [9]. Refractory anti-SRP myopathy demonstrated marked and sustained clinical response to the combination of prednisone, plasma exchange, and repeated courses of Rituximab [10].
We present a clinical case of patient with severe necrotizing myopathy with positive anti-SRP autoantibodies.
A 36-year-old male patient presented in the rheumatology clinic in October 2019 with complaints of progressive weakness and fatigue of the muscles in upper and lower limbs, unstable gait, palpitations. Weakness was characterized by difficulty standing up from sitting and squatting position, combing hair and changing cloths. The weakness in the extremities had a progressive course. Our patient was not taking any myotoxic drugs or statins. Family history for any disease was negative.
Physical exam findings were severely weak upper- and lower extremity proximal and distal muscle groups with atrophy of the quadriceps and gluteus muscles. There were no skin rashes, oral ulcers, photosensitivity, and hair fall or weight loss. Manual muscle strength was graded 1/5 in the proximal lower extremities and 2/5 in the proximal upper extremities. Physical examinations of respiratory and gastrointestinal symptoms were unremarkable. It was found irregular heart rhythm. Liver, spleen, peripheral lymph nodes were not enlarged.
The laboratory studies revealed normal non-specific inflammatory markers (erythrocyte sedimentation rate ESR 7 mm/h, C-reactive protein 0.7 mg/l (normal < 6 mg/l)). Other investigations showed haemoglobin 170 g/l, total leukocyte count 7.77 × 109/l, thrombocytes 202 × 109/l. Renal function rest results were normal. Liver enzymes were increased (ASAT 217 U/l (normal < 40 U/l); ALAT 225 U/l (normal < 41 U/l). CK and LDH levels were markedly elevated, respectively 8072 U/l (normal range 24-195 U/l) and 1550 U/l (normal < 460 U/l). Urine test results were normal. Hepatitis B and C, HIV, were all negative.
Electromyography was characterized by the presence of polyphasic, short-duration, low-amplitude MUAPs (motor unit action potentials) in the lower and upper limbs. Motor response from the right tibial nerve to gastrocnemius muscle was very low, as registered single motor units. Other motor and sensory nerves were normal.
24-hour Holter monitoring showed unstable sinus heart rhythm, heart rate was between 54-115 per minute. Supraventricular premature beats with couplets and triplets, episode of supraventricular tachycardia, grouping ventricular bigeminy and trigeminy were registered on Holter ECG (Figure 1).
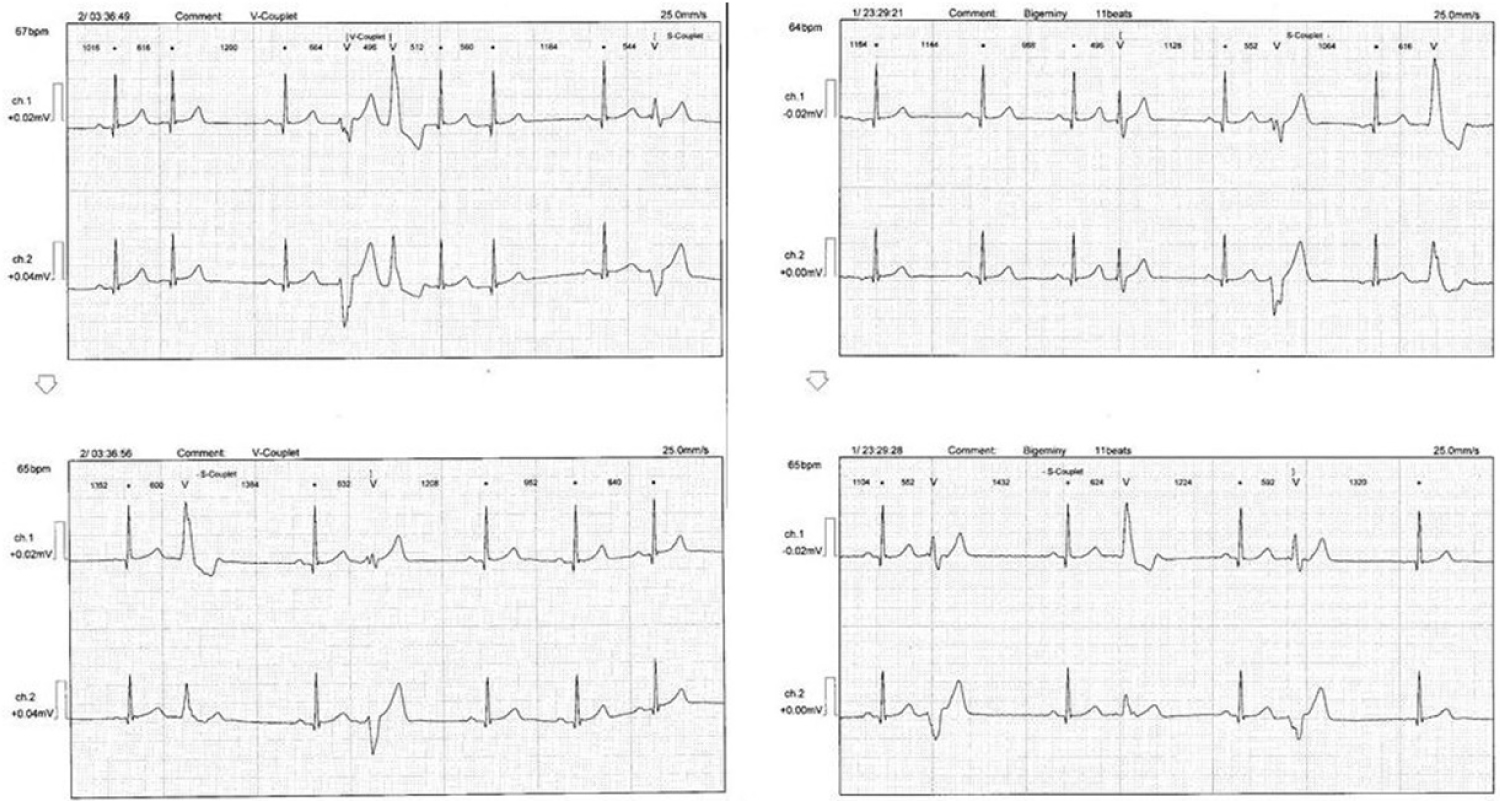 Figure 1: Supraventricular and ventricular extrasystoles. View Figure 1
Figure 1: Supraventricular and ventricular extrasystoles. View Figure 1
Echocardiography showed normal chamber size and left ventricular ejection fraction. A computed tomography (CT) scan of the chest and abdomen was performed to screen for any possible malignancy leading to myositis. The result of CT scan was negative for any malignancy. Magnetic resonance tomography of the proximal muscles of lower limbs demonstrated muscle oedema of the posterior compartments (Figure 2).
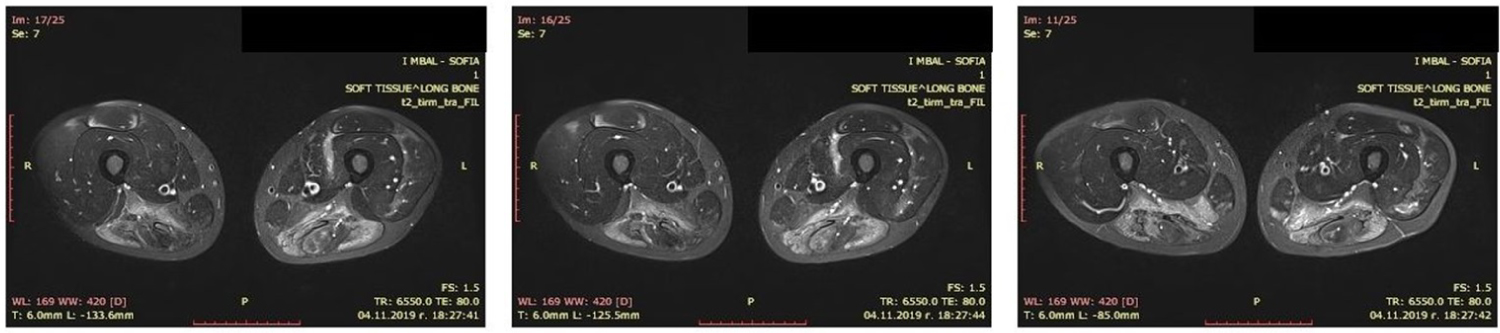 Figure 2: Thigh muscle MRI showed muscle oedema of the posterior proximal muscles. View Figure 2
Figure 2: Thigh muscle MRI showed muscle oedema of the posterior proximal muscles. View Figure 2
The specified tests antinuclear antibodies on indirect immunofluorescence were positive ANA 1:1280 (normal < 1:160). From extractible nuclear antigens, Abs against ribosome P antigen were positive. Myositis-specific Abs were tested, as the patient was positive for anti-SRP antibodies.
Muscle biopsy revealed necrotizing changes without any evidence of significant inflammatory process and scattered muscle fibre regeneration (Figure 3).
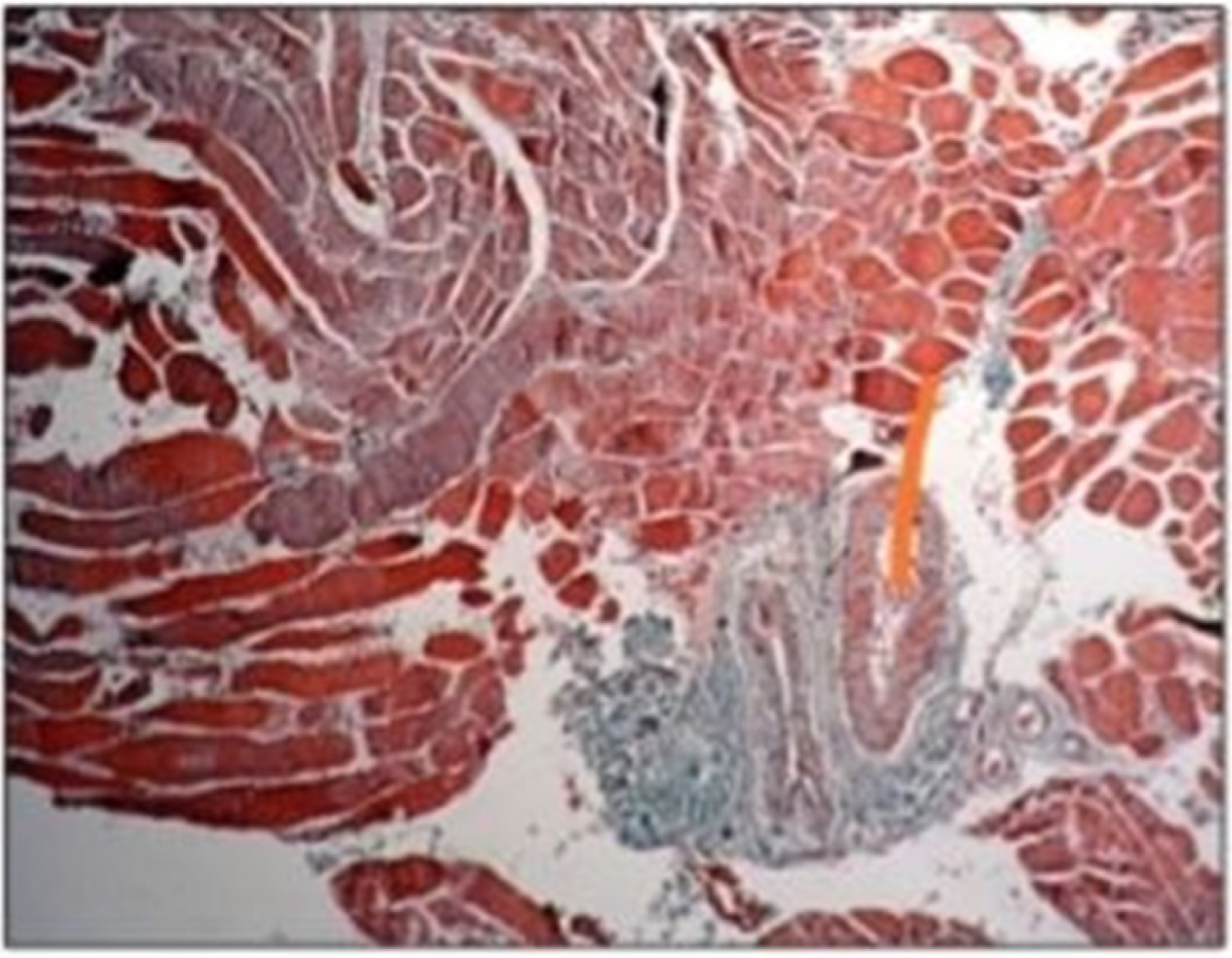 Figure 3: Anisometry, necrotic altered rhabdomyocytes, fibrosing rhambomyocytes, perivascular fibrosis. View Figure 3
Figure 3: Anisometry, necrotic altered rhabdomyocytes, fibrosing rhambomyocytes, perivascular fibrosis. View Figure 3
The following differential diagnosis was discussed:
• Idiopathic inflammatory myopathy (myositis)
• Limb-girdle muscular dystrophy
Myositis and limb-girdle muscular dystrophy are characterized by progressive muscle wasting which affect predominantly hip and shoulder muscles. Both diseases occur with elevated CK serum levels. Electromyographic pattern in myositis and limb girdle muscular dystrophy is presented by polyphasic, short-duration and low amplitude MUAPs. The differentiation of the two diseases is based on the result of histopathological examination, Abs and the result of molecular genetic testing. Histopathological result in myositis is presented by anisometry, mononuclear cell infiltrates, muscle fiber necrosis, myophagy, fiber degeneration. Muscle biopsy in limb-girdle dystrophy shows "dystrophic" triad - anisometry, muscle fiber necrosis and interstitial fibrosis. Necrotic altered rhabdomyocytes dominated in muscle biopsy in the present clinical case. The result of the muscle biopsy did not give a definite answer whether it was myositis or limb girdle muscular dystrophy.
After proving the positive anti-SRP Abs and negative result of the molecular genetic test, a diagnosis was made - necrotizing myopathy with positive anti-SRP Abs.
Pulse therapy with methylprednisolone 1000 mg intravenous in three consecutive days was applied for three months. Between the pulses, therapy with oral steroids (methylprednisolone) 60 mg/day was started. Azathioprine 100 mg/day was added to his treatment regimen. Pulse therapy with intravenous immunoglobulin (1 gr/kg i.v.) was applied monthly for three months. The patient received four plasmapheresis treatments. Three months later, the CK, LDH, ASAT, ALAT normalized with concurrent improvement of the muscle weakness and persisting muscle atrophy. The methylprednisolone dosage between the pulses was tapered slowly, Azathioprine and intravenous immunoglobulin were continued.
Immune-mediated necrotizing myopathy is a relatively new disease, described for the first time in 2004. There have been no randomized trials and except for a few anecdotal reports, there are no large enough case series available to make definite conclusions or formal recommendations. IMNM is a form of myositis since it is an inflammatory myopathy with strict involvement of the skeletal muscles albeit lymphocytic infiltration is sparse or absent. The clinical picture, pathological features and autoantibodies of IMNM differ markedly from those of antisynthetase syndrome and non-specific myositis. Anti-HMGCR and anti-SRP Abs are strongly specific for IMNM, but their role in the onset and/or persistence of disease is unknown [11].
Muscle weakness in IMNM is the predominant clinical feature and affects the lower limbs most severely [12]. In addition, dysphagia is frequently found in these patients [13,14]. Anti-SRP myositis patients may also have cardiac involvement, including rhythm or conduction abnormalities as well as cardiac insufficiency [13,14]. Other extra-muscular manifestations may include interstitial lung disease but is typically mild. Importantly, the outcome of anti-SRP myopathy is poor since only half of patients recover normal strength after four years [15]. Yves Allenbach compared clinical phenotypes of both anti-SRP and anti-HMGCR patients, showing that anti-SRP myopathy is usually more severe and with more intense muscle damage [11].
We present a clinical case of necrotizing autoimmune myopathy with positive anti-SRP Abs with typical presentation. Patient manifested with proximal muscle weakness with atrophy of the quadriceps and gluteus muscles, conduction abnormalities, elevated CK levels, and myopathic EMG findings. Muscle biopsy demonstrated prominent necrotic myofibres. IMNMs with positive anti-SRP Abs may mimic muscular dystrophy. Therefore we discussed differential diagnosis between autoimmune myopathy and limb-girdle muscle dystrophy in the present case. In both, myositis and limb-girdle muscular dystrophy, muscle weakness has chronic progressive course and CK levels are increased. In anti-SRP patients' muscle biopsies, necrotic muscle fibres are distributed with a diffuse pattern, lymphocytic infiltration is sparse or absent, and muscle fibre regeneration is scattered [11]. Most dystrophies are characterized by fibre-size variation, which is more prominent as compared to IMNMs, and small regenerating fibres are mostly clustering in contrast to IMNM [11]. The positive autoantibodies against signal recognition particle (anti-SRP Abs) and the result of muscle biopsy had key role in making of diagnosis in the current case. The etiology of IMNM is unknown, some of the risk factors include statin use, malignancy, a human immunodeficiency virus infection, or connective tissue disease [3,16,17]. The patient hadn't a history of using statin. We excluded malignancy, as other connective tissue disease.
First line treatment remains corticosteroids, high dose prednisone/prednisolone or pulsed intravenous methylprednisolone [11]. Some experts are recommending intravenous immunoglobulin as a first line [18]. If a prolonged course of corticosteroids is anticipated, or the patients' therapy cannot be lowered to less than 10 mg/day, or if they have concomitant medical issues such as interstitial lung disease, osteoporosis, or diabetes, it is recommended to add a second agent. Azathioprine is the preferred agent with a dose of 3 mg/kg. Methotrexate can also be used at 20-25 mg per week orally or subcutaneously. Third line drugs would include rituximab. There has been considerable experience with rituximab in SRP autoantibody associated myositis and anti-synthetase syndrome. Some experts created consensus for the initial treatment of IMNM [11].
Early administration of therapy of corticosteroids with immunosuppressant, within three months, was found to be beneficial [3,13]. According to Kassardjian, et al. the early initiation of IVIG was seen to be advantageous [19]. In the current case, we started combined therapy with high dose corticosteroids, Azathioprine, IVIG and plasmapheresis. J Arlet, et al. demonstrated marked and sustained clinical response to the combination of prednisone, plasma exchange, and repeated courses of Rituximab in two patients with refractory anti-SRP myopathy [10]. Three months later, the muscle enzymes normalized with concurrent improvement of the muscle weakness, persisting muscle atrophy and persisting arrhythmia. After the sixth month, in case of insufficient effect of the therapy, treatment with Rituximab will be started, as a third line of treatment.
In summary, SRP myopathy is one of the most disabling auto-immune myopathies, characterized by an initially severe muscle weakness as well as often poor muscle recovery even with treatment. Therefore it is necessary to apply aggressive combined therapy. In brief, initial treatment for SRP myopathy usually includes corticosteroids and immunosuppression agent, added at the same time or within one month (depending on severity and response to treatment). For severe cases of SRP myopathy, rituximab should always be added within six months if other strategies are failing.