Adrenal tumors may be a benign finding or imply a high level of morbidity and mortality due to their hormonal activity, or to a possible malignant histology. Medical literature indicates that this condition is due to the improvement of the technique and dissemination in a wide range of imaging methods, increasing considerably the diagnosis of adrenal nodules and its immediate treatment. When treating a patient with adrenal tumors, the main concern of the professional should be to establish if the lesion is a malignant neoplasm and if there is some hormonal functioning, which are two occurrences that usually require surgery. To establish if a lesion is hormonally working it is necessary to perform complete clinical and endocrine assessments. Thus, it should be part of the routine of the general practitioner to recognize and evaluate whether the lesion is malignant or functionant, situations in which adrenalectomy is necessary.
Adrenocortical tumors, Adenoma, Adrenal, Cushing's syndrome, Aldosteronoma
ACTH: Adrenocorticotropic Hormone; DEXA: Dexamethasone; FEO: Pheochromocytoma; FEO/ PGL: Feocromocitoma/Paraganglioma; HA: Systemic Arterial Hypertension; SAGH: Autonomous Subclinical Hypersecretion of Glucocorticoids; SC: Serum Cortisol; UFC: Urine Free Cortisol; IA: Adrenal Incidentaloma; HU: Hounsfield Unit
Adrenal gland tumors are common diseases in clinical practices. According to the way they manifest themselves, they are classified into functionants (which produce hormones) and non-functioning (also known as mufflers). In terms of biological behaviour, they can be divided into benign or malignant tumors [1].
Most adrenocortical tumors are benign, unilateral and non-functioning adenomas, presenting less than 4 cm in diameter, perceived during abdominal imaging studies [1].
This "incidental finding" is given the name of adrenal incidentaloma and is defined as an adrenal mass, clinically unsuspected, which is found in imaging studies conducted for other reasons than the evaluation of the adrenal glands. In recent years, the prevalence of adrenal adenomas has increased due to the use of abdominal imaging with increasing sensitivity [2].
Functioning adrenal tumors are usually of the benign adenoma type, which causes, for example, Cushing's syndrome, primary aldosteronism or, not so commonly, virilization [3,4]. In the bone marrow, pheochromocytomas, secretory tumors of catecholamines, which stand out despite being rare and imply in great morbidity and mortality [1].
In a combined study with a different selection and diagnosis criteria, we had the main etiologies of adrenal tumors: Adenomas 41%, Metastases 19%, Carcinomas 10%, Myelolipomas 9% and Pheochromocytomas 8%. Most of the remaining cases (13%) were benign lesions, such as adrenal cysts [2].
In clinical reports, adrenal incidentalomas show peak incidence in the fifth to seventh decades. The mean age of patients at diagnosis is 55 years, with no significant differences in age between female and male [5].
The prevalence increases with age; the rate is less than 1% for patients under 30 and 7% for patients aged 70 and over [6].
Attributed to hormonal production, although most tumors are not functioning, a slightly increased production of certain hormones can be verified in at least 15% of the cases.
Adenomas or adrenal carcinomas secreting cortisol, pheochromocytomas, aldosteronomas and secretory lesions of androgens are the types of secretory or functionant adrenal masses diagnosed more recurrently [3].
These tumors usually produce decreased amounts of cortisol, which, in most cases, are not sufficient to increase the excretion of free cortisol in urine. They are, however, capable of causing the suppression of the hypothalamic-pituitary axis. Usually, there are no manifestations related to Cushing's syndrome in patients. For this reason, this condition is known as subclinical Cushing's syndrome or subclinical hypercortisolism [3].
It is important to highlight the difference between subclinical Cushing's syndrome, characterized by a clinically non-manifested biochemical abnormality and pre-clinical Cushing's syndrome, which is the initial stage of the development of the syndrome itself. The autonomous subclinical hypersecretion of glucocorticoids (SAGH) is the most current term, proposed to define an autonomous cortisol secretion by an adrenal adenoma in patients without symptoms of Cushing's Syndrome [2].
In adults, the most suggestive signs and symptoms of the presence of hypercortisolism include proximal muscle weakness, facial plethora, loss of limbs with increased fat in the abdomen and face, wide purple streaks, hematomas without obvious trauma and supraclavicular compresses [7].
However, due to many symptoms of hypercortisolism, among them, hypertension and diabetes are not characteristic, and the degree of their clinical appearance is consistent with the variation in the extent of hormonal overproduction, the precise indication of the prevalence of SAGH, will be subjected to the results obtained by the methods of the tests used and the criteria for the selection of symptomatic patients for the confirmation of the disease [2].
When you suspect Cushing's syndrome, a 1 mg dexamethasone suppression test should be performed overnight. The day before the collection of serum cortisol that occurs at 8 a.m., the patient takes 1 mg of dexamethasone orally at 11 pm [3].
It is considered an abnormal cortisol response when it is presented in the range of 1.8 to 5.0 mcg/dl, or more specifically:
A) If < 1.8 mcg/dL: The autonomous production of cortisol can practically be excluded;
B) If between 1.8 and 5 mcg/dL: Indeterminate results;
C) > 5 mcg/dL: Highly probable diagnosis of Cushing's syndrome;
An abnormal suppression with dexamethasone is equivalent to a suspicious screening, thus it should be confirmed by dosage of free urinary cortisol of 24 hours, after administration of 8 mg dexamethasone overnight and serum ACTH dosage, so that it can be determined the origin of Cushing's syndrome, which usually shows up with unsuppressed levels (ACTH-dependent) [3].
In the case of the diagnosis of subclinical Cushing's syndrome, the following criteria is used:
-Plasmatic Cortisol > 5 mcg/dL in the 1 mg dexamethasone test without any other stigma or at least 2 of the following results [3].
1. Plasma ACTH < 10 pg/ml;
2. Free urinary Cortisol in a sample of 24 hours increased;
3. Serum Cortisol > 3 mcg/dL in the 1 mg dexamethasone test (Figure 1).
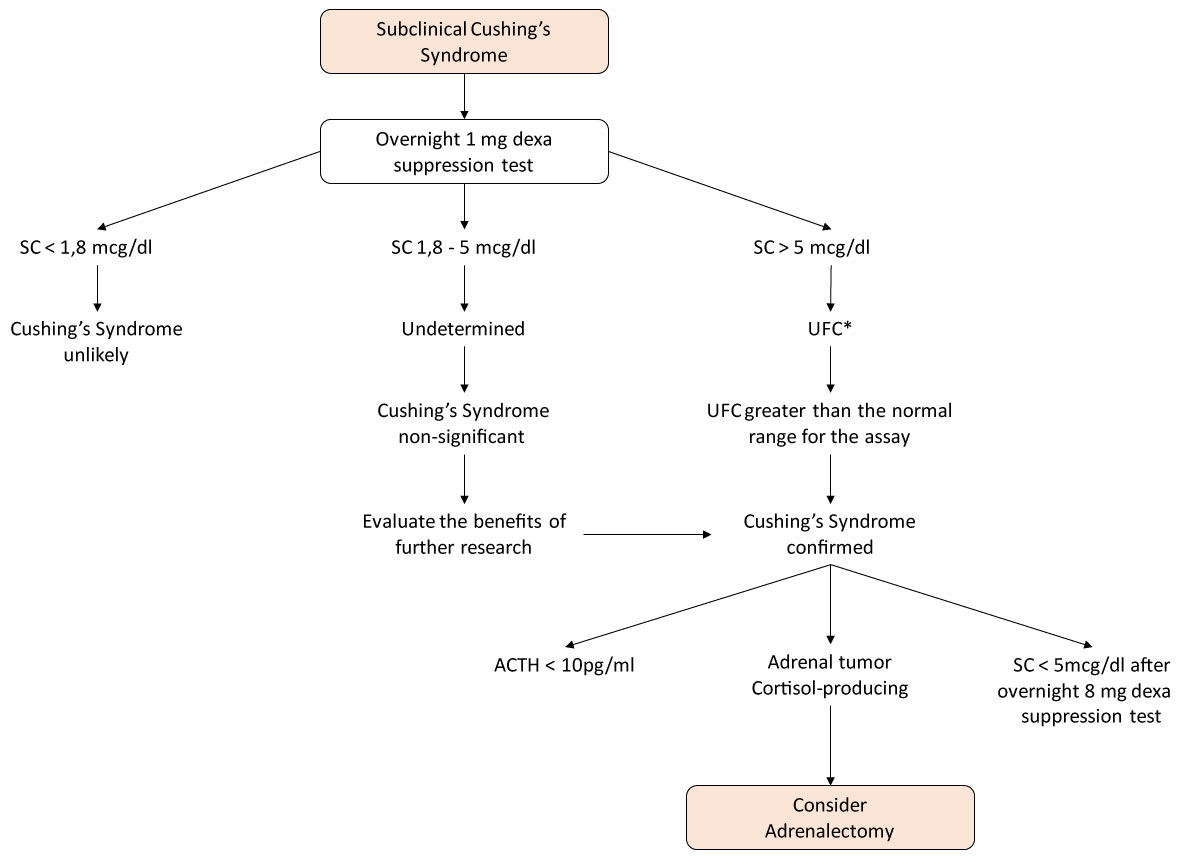 Figure 1: Algorithm for investigating Cushing's syndrome in Adrenals Incidentalomas. *At least two measurements.
View Figure 1
Figure 1: Algorithm for investigating Cushing's syndrome in Adrenals Incidentalomas. *At least two measurements.
View Figure 1
Thus, it is difficult to reach a consensus for the approach of Subclinical Cushing's syndrome, which can be treated clinically or through surgery [4]. For patients with many comorbidities that may be related to hypercortisolism, the risk/benefit of adrenalectomy should be considered as a treatment. A large part of the patients undergoing this surgery may develop acute Adrenal insufficiency (sometimes fatal), so it is extremely important to perioperative coverage with glucocorticoid administration.
Pheochromocytomas are tumors of chromaffin cells of the adrenal medulla that produce, store, metabolized and secrete catecholamines, in some cases, other peptide hormones paragangliomas (PGL) are similar tumors, but of extra origin. Feo/PGL syndrome is a rare disease, with an estimated prevalence between 0.1 and 0.2% of the population of hypertensive individuals [3].
Most catecholamine secreting tumors are sporadic. However, some patients (approximately 40%) have the disease as part of a family disorder; in these patients, catecholamine secreting tumors are more likely to be bilateral adrenal pheochromocytomas or paragangliomas.
There are several family disorders associated with pheochromocytoma, all with autosomal dominant inheritance:
• Von Hippel-Lindau syndrome (VHL), associated with mutations in the VHL tumor suppressor gene.
• Endocrine multiple neoplasm type 2 (MEN2), associated with mutations in proto-oncogene RET.
• Pheochromocytoma is also observed, although infrequently, of neurofibromatosis type 1 (NF1), due to mutations in the NF1 gene.
• The approximate frequency of pheochromocytoma in these disorders is 10 to 20% in VHL syndrome, 50% in MEN2 and 3% in NF1 [8].
These tumors are of particular importance because, although rare (as well as the secretory adenomas of aldosterone) give rise to a surgically correctable form of hypertension [9]. Hypertension is often paroxysmal. The classic triad of symptoms in patients with pheochromocytoma consists of episodic headaches, sweating and tachycardia, and there is a prevalence of patients who do not present the three classic symptoms. Besides the patients with the typical clinical manifestations of the disease, the investigation should also be done in individuals with a family history of FEO or medullary thyroid carcinoma, in the presence of HA in young, HA of difficult control or anesthetic induction.
The diagnosis of pheochromocytoma is typically made by measurements of metanephrines and fracted catecholamines in the urine and plasma, as shown in Figure 2 [10].
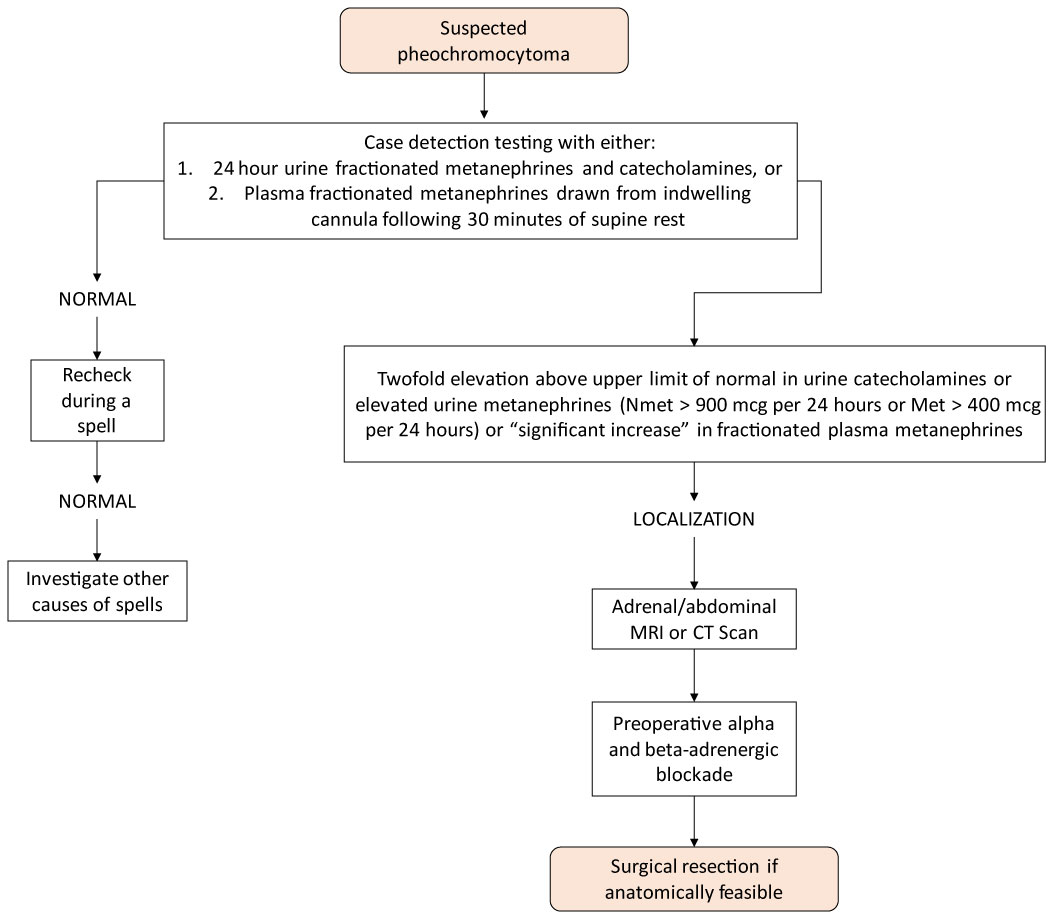 Figure 2: Algorithm for the investigation of Pheochromocytomas in Adrenals Incidentalomas.
View Figure 2
Figure 2: Algorithm for the investigation of Pheochromocytomas in Adrenals Incidentalomas.
View Figure 2
As for the image methods, Pheochromocytomas has greater attenuation on unenhanced CT (> 20 HU), increased vascularization of the mass, delay in contrast medium washing (10 minutes after contrast administration, an absolute contrast medium wash of less than 50%), high signal strength on T2-weighted MRI, cystic and hemorrhagic changes and variable size and can be bilateral.
In contrast, the imaging characteristics that suggest adrenal carcinoma or metastases include: Irregular shape, inhomogeneous density, high unenhanced CT attenuation values (> 20 HU), delayed contrast medium washout (eg, < 50 percent at 10 minutes), diameter > 4 cm, and tumor calcification [11].
Aldosteronomas are uncommon and difficult to detect and are clinically characterized by systemic arterial hypertension related to hypokalemia. Considering that most patients are asymptomatic regarding hypocalcemia, that is, they are normocalemic, it is recommended that all patients with hypertension associated with adrenal incidosis be evaluated by measuring their plasma concentration of Aldosterone and renin Plasma activity and the ratio between them [3,12] that is greater than 30 is highly suggestive of autonomous aldosterone production. A greater reason than 50 clearly distinguishes primary aldosteronism from other forms of essential hypertension [2,13-15] already a result less than 20 disputes the diagnosis and results in the interval between 20 to 30, indicate a more accurate diagnosis.
Patients who use spironolactone cannot be evaluated by the aldosterone relationship and plasma activity. Other medications that may contribute to doubtful results are beta-blockers, central alpha-adrenergic agonist and anti-inflammatories. Similarly, some drugs may show a reduction in the inhibitor of the conservative angiotensin enzyme, such as aldosterone receptor blocker, thiazine inhibitors and dihydropyridine of the calcium channel Figure 3 [4].
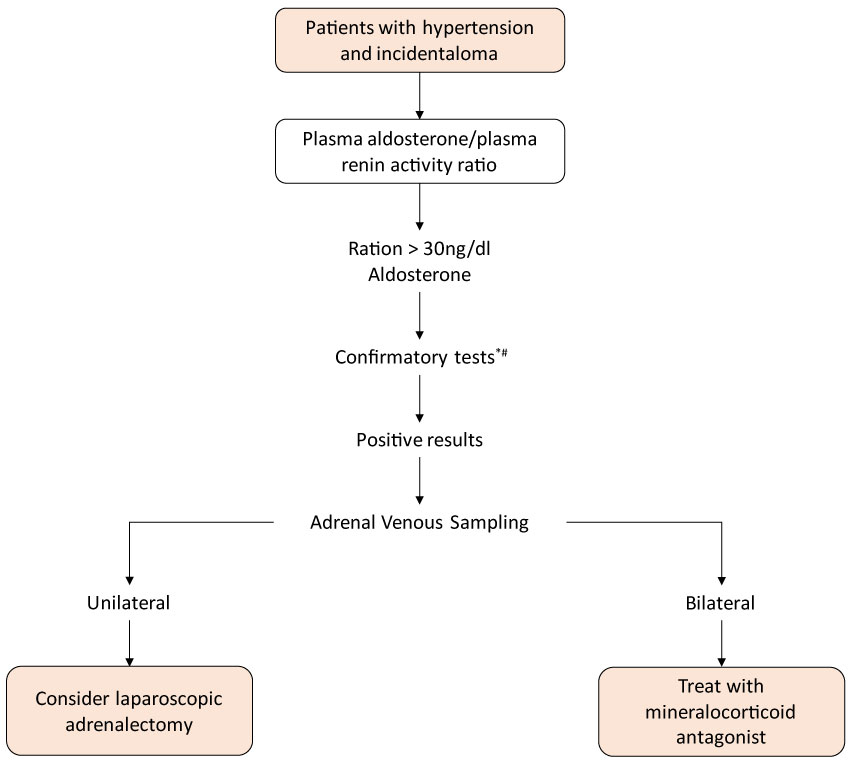 Figure 3: Algorithm for the investigation of Hyperaldosteronism in Adrenals Incidentalomas. *Most commonly used Confirmatory tests: Oral sodium load test, intravenous saline infusion test.
View Figure 3
Figure 3: Algorithm for the investigation of Hyperaldosteronism in Adrenals Incidentalomas. *Most commonly used Confirmatory tests: Oral sodium load test, intravenous saline infusion test.
View Figure 3
Adrenal catheterization is the method of evaluation conducive to verifying whether the increase in aldosterone production in patients over 40 years, with confirmed Hyperaldosteronism, is actually caused by incidentaloma or adrenal hyperplasia.
In such cases, adrenalectomy would not solve hormonal hyperproduction, which should be kept under control with the use of medication, aldosterone antagonists such as spironolactone [4].
In patients with spontaneous hypokalemia, plasma renin below detection levels plus plasma aldosterone > 20 ng/dl, it is suggested that there is no need for further confirmatory testing.
In cases of congenital adrenal hyperplasia due to a 21-hydroxylase deficiency, it is common to find adrenal masses, uni or bilateral, as a consequence of the chronic excessive stimulation of adrenals by ACTH [3].
The sex hormone that produces adrenals Adenomas is very rare. Androgen-producing Carcinomas are also uncommon. Cases of androgens or excess estrogen are rarely described in patients with benign adrenocortical adenomas, but, in general, they are manifested by symptoms or signs of virilization in women (acne, hirsutism) or feminization in men (Gynecomastia). Thus, such lesions cannot be considered as true IAs. Therefore, the need to measure sex hormones and steroid precursors is limited in cases of adrenal lesions with indeterminacy or suspicion of imaging features of malignancy, where elevated levels may point to the adrenocortical origin of the tumor and suggesting the presence of an adrenal Adenocarcinoma [16]. Adrenalectomy is indicated for the control of hormones in individuals with virilization or high concentrations of androgens [3].
Estrogen-producing tumors are uncommon and, in most cases, malignant. The presence of these tumors in men is usually manifested through feminization with gynecomastia, decreased libido, atrophy of the testicles; in women, it can manifest through breast tenderness and bleeding [12]. In such cases, adrenalectomy can also be indicated.
The investigative studies of adrenals imaging are of particular relevance to the understanding that the professional of this area is attentive and curious about any trait of abnormality that suspects the health of the adrenal glands.
In parallel, we can observe the importance of knowledge of the effective methods for diagnosis and for the treatment pertinent to each case detected.
Of all the protocols used and proposed for the hormonal investigation, the one with the highest recurrence is the one that uses serum cortisol after nocturnal suspension, with 1 mg dexamethasone (for subclinical hypercortisolism) and Plasma metanephrines (for the FEO). The investigation of Aldosteronomas is only indicated for cases of hypertension and or hypokalemia, the determination of plasma aldosterone and plasma renin activity.
Numerous studies published and disseminated in medical symposia mention several protocols used for hormonal investigation, the observation and discovery of abnormality of tissue cells of the adrenal glands, the different tumors that these Glands can shelter and preventive and treatment measures of diseases, offering quality of life to patients.
None of the authors have a conflict of interest.