The interaction between CD8 and HLA class I is largely monomorphic and of low affinity. Previous HLA mutation studies have indicated that manipulation of the affinity of this interaction can have dramatic biological effects. Reductions in CD8-HLA class I affinity can lead to inhibition of T cell mediated target cell killing, whilst modest affinity increases lead to enhanced target cell killing whilst retaining antigenic specificity. Here it is shown that target cells bearing a high affinity chimeric MHC molecule termed a 'Supertarget' comprising the HLA-A2 molecule incorporating the murine MHC Kb alpha 3 domain can be killed by T cells irrespective of their native HLA or peptide specificity.
We will review the immunotherapy potential of this Supertarget protein and also how novel drugs acting as agonists or antagonists at the CD8-HLA class I interface may be of clinical value.
T cell, Cancer, Autoimmunity, Immunology, Protein-protein interaction
The use of the cellular immune system to treat cancer is one of the most rapidly evolving areas of therapy. Historically, T cell-based therapies aimed to boost the natural response of the immune system to tumours cells, using relatively simple technology including cytokines and cancer vaccines [1,2]. More recently more complex technology has been applied to manipulate the cellular immune system.
One approach has been to enhance the native T cell response to tumours, by reducing the natural inhibitory controls of the cellular immune system rather than delivering direct stimulation. Two approaches to this are now in routine clinical practice using monoclonal antibodies that block either the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) system or the programmed cell death protein 1- programmed death-ligand 1 (PD1-PDL-1) interaction [3]. Both of these therapeutic approaches deliver considerable benefit to patients over an increasingly wide range of cancers including melanoma, lung cancer, renal cancer, Hodgkin's lymphoma and bladder cancer [4-6]. It appears likely that malignancies with high mutational burdens such as melanoma and lung are likely to respond most effectively to these therapies [7,8].
However, the nature of how T cells recognise tumour cells, via the T cell receptor (TCR) interacting with the HLA class I molecule displaying an antigenic peptide of tumour origin, may limit the potential efficacy of cellular immunotherapy [9,10]. Typically, these interactions are of low affinity, therefore the resultant strength and specificity of the immune response against tumour cells may remain modest even with the blockade of co-inhibitory receptor pathways. This may in part explain the proportion of patients that currently do not respond to these therapies.
A second therapeutic approach is to retarget cytotoxic T cells to attack tumour cells using an alternate recognition system aside from the natural TCR/HLA class I peptide interaction. Technologies with this group include; bi-specific antibody fragments, including Blinitumomab [11], which has shown efficacy in leukemia and lymphoma [12,13], chimeric antigen receptor T cells (CAR-T cells) that express a recombinant receptor on the surface of transfected T cells [14] and immune mobilising monoclonal T-cell receptors against cancer (ImmTacs), which target tumour cells via a high affinity engineered T cell receptor fragments [15]. However, these systems are complex, expensive and may also have practical limitations to their activity and widespread clinical utility. An alternate approach to these strategies is to change the immunological appearance of the tumour cell so that it appears to mimic a virally infected cell, with the aim of interacting with the high levels of naturally occurring virus specific T cells that can subsequently kill the tumour cell. This aim can be accomplished by delivering recombinant HLA class I molecules which contain a viral peptide to the surface of tumour cells using a monoclonal antibody delivery system.
We originally described this concept using a simple biotin-streptavidin based system. This work indicated that recombinant HLA class I peptide complexes when attached to the surface of a tumour cells serve as an efficient target for T cells specific for the HLA class I/peptide complex and result in effective tumour cell killing [16, 17]. The technology has been subsequently developed in both the commercial and academic setting using a number of differing antibody delivery systems including, chemical conjugates, HLA-sfv fusion proteins, HLA-IgG fusions and the antibody dock and lock technology [18-20].
Each of these systems indicates that effective target cell killing can be produced in vitro and in vivo using re-directed virus specific T cells as the effectors. A more recent publication from the team at Roche gives a good summary of the potential of the technology [21]. However, whilst each of these delivery systems appear to offer effective in vitro and in vivo target cell killing the physiological restriction of the immune response to a single HLA allele/peptide may limit both the efficacy and the wider applicability of the system.
Here we discuss how engineering changes to the protein structure of the HLA class I molecule can dramatically alter the affinity of the interaction with the CD8 coreceptor expressed at the T cell surface and how the resultant changes in T cell activation and even specificity could potentially be exploited via this and other therapeutic approaches.
CD8 is a transmembrane glycoprotein of the immunoglobulin superfamily that is expressed on the surface of cytotoxic T cells. The cell surface molecule can be present as either an alpha-alpha homodimer or alpha-beta heterodimer. The alpha and beta chains of CD8 are similar, in that they both have a short cytoplasmic region, a single transmembrane domain, a glycosylated stalk and an immunoglobulin like extra-cellular domain [22].
The alpha-beta CD8 heterodimer is expressed only on conventional HLA class I restricted alpha beta TCR+ cytotoxic T cells. In contrast, the CD8 alpha-alpha homodimer, whilst present in low levels on CD8 T cells, is also expressed on additional cell subsets such as gamma delta T cells and NK cells [23,24].
The main interaction between CD8 and the HLA class I molecule occurs via a short length of monomorphic amino acids on the alpha 3 domain of the HLA molecule [25]. At this point two complementary determining region (CDR) like loops of the CD8 molecule interact with an area encompassing residues 223-227 of the HLA class I alpha 3 domain as shown in Figure 1.
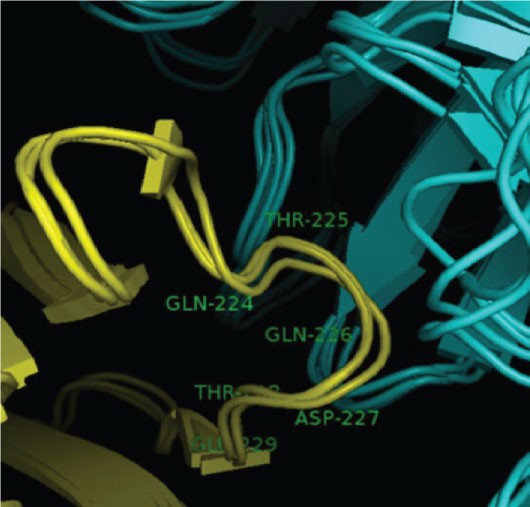 Figure 1: The interaction between the wild type HLA class I alpha 3 loop, residues 224-229 (yellow) and CD8 (blue). Only the path of the protein backbone is shown in cartoon representation, for clarity.
View Figure 1
Figure 1: The interaction between the wild type HLA class I alpha 3 loop, residues 224-229 (yellow) and CD8 (blue). Only the path of the protein backbone is shown in cartoon representation, for clarity.
View Figure 1
The native CD8-HLA class I interaction is characterized by a relatively low affinity (KD = 145 µM) and rapid kinetics [26-28]. This contrasts with the generally higher affinity of the interaction (KD = 1-10 µM) seen between the TCR and HLA class I/peptide complex [29].
Despite the low affinity, the binding of the HLA class I molecule to CD8 at the cell surface enhances the association rate of HLA class I complexes with TCRs and increases the interaction half-life [30]. In addition to stabilising the TCR/peptide-HLA interaction, the binding of CD8 leads to recruitment of p56lck and triggers signalling pathways downstream of the TCR/HLA triggering [31].
Previous in vitro studies using point mutation changes to the HLA class I alpha 3 domain that alter the binding affinity of the CD8-HLA class I interaction, have indicated that if this low affinity interaction is altered then there can be a very significant change to the T cell activity. Abrogation of this interaction via an amino acid substitution (D227K/T228A) in the alpha 3 domain of the HLA class I molecule leads to a reduction in the binding affinity to KD > 10,000 µM. This change leads to a dramatic reduction in production of cytokines and lytic ability of T cells on contact with their target cells as shown in Figure 2.
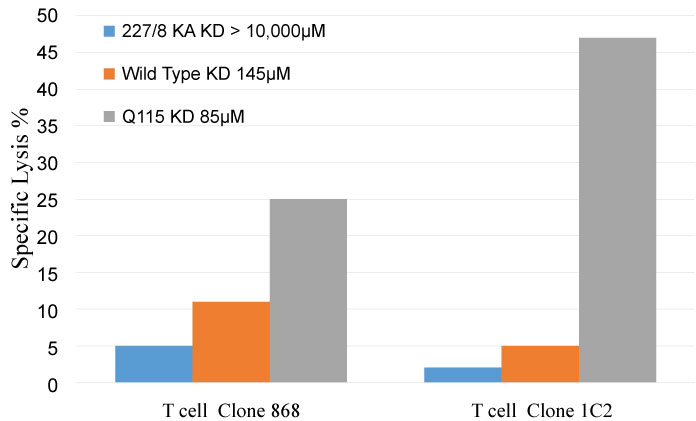 Figure 2: The impact of changes in CD8-HLA class I binding affinities on T cell allele/peptide specific lytic function. The ability of two HLA-A2 specific T cell lines (868 and 1C2) to produce cell lysis of peptide pulsed C1R-A2 bearing HLA-A2 molecules of differing affinities. 227/8KA (> 10,000 µM), Wildtype (145 µM) and Q115 (85 µM). Data from reference (32) (E:T 2:1, 2 hour incubation).
View Figure 2
Figure 2: The impact of changes in CD8-HLA class I binding affinities on T cell allele/peptide specific lytic function. The ability of two HLA-A2 specific T cell lines (868 and 1C2) to produce cell lysis of peptide pulsed C1R-A2 bearing HLA-A2 molecules of differing affinities. 227/8KA (> 10,000 µM), Wildtype (145 µM) and Q115 (85 µM). Data from reference (32) (E:T 2:1, 2 hour incubation).
View Figure 2
In contrast protein engineered changes (Q115E) that moderately increase the native affinity of the CD8-HLA class I interaction to KD ~85 µM result in enhancement of T cell activity compared to the wild type HLA-A2 as shown in Figure 2. However, whilst the strength of the T cell immune response to cells bearing the appropriate HLA class I/peptide complex is significantly enhanced by this modest change in CD8-HLA class I affinity, the T cell -target cell specificity is unchanged [32].
When the affinity of the CD8-HLA class I interaction is increased further to KD ~10 µM, by engineering a chimeric HLA-A2/Kb molecule, that bears the murine MHC alpha 3 domain, there is a dramatic change in the specificity of the T cell interaction. In this situation, there is loss of T cell HLA/peptide specificity and T cells of any specificity can be activated via this interaction with the enhanced HLA molecule irrespective of the identity of the presented peptide [33].
Whilst to date, the published data on this phenomenon has focused on the activation and expansion of T cells, our recent data indicates that target cells bearing HLA-A2/Kb complexes can be killed by CD8 T cells irrespective of their cognate HLA or peptide specificity. The data in Figure 3a indicates that T cell clones with specificity for either HLA-A2 or HLA-B35 can effective kill A2/Kb targets, whilst in Figure 3b purified peripheral blood CD8 cells from A2+ve and A2-ve donors both also produce effective killing of the target cells bearing HLA-A2/Kb.
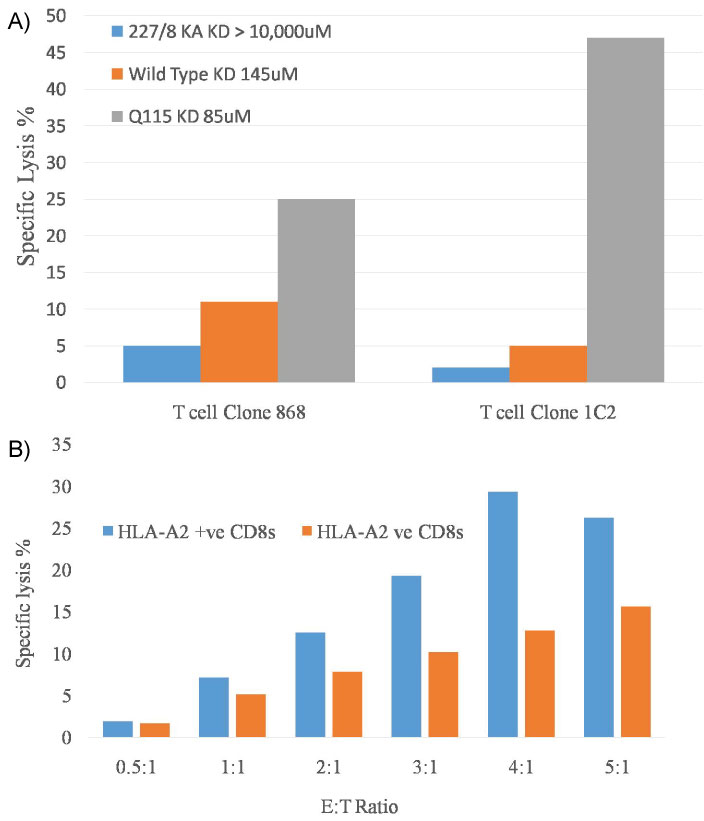 Figure 3: a,b) HLA class I unrestricted T cell clone mediated lysis of C1R B cells expressing HLA-A2/Kb.
Figure 3: a,b) HLA class I unrestricted T cell clone mediated lysis of C1R B cells expressing HLA-A2/Kb.
For these FATAL assays 3 × 104 eF670+ HLA-A2/Kb+ C1Rs and control eF670- HLA-A2wt+ C1Rs cells were combined with CD8+ T-cells at varying (E:T) ratios: Assays containing no CD8+ effector T-cells were used as a baseline to measure background cell death. Following a 12-hour incubation period (37 ℃, 0.5% CO2), the cells were harvested, stained with anti-CD8-BV785 and the viability dye Aqua, prior to data acquisition on a Fortessa flow cytometer (BD Biosciences).
Specific lysis of HLA-A2Kb-expressing targets by HLA-A2 and non-HLA-A2 restricted CD8+ T-cell clones MelC5 (specific for HLA-A2/melan-A) and SB14 (specific for HLA-B35/EBNA-1) are shown in Figure 3a.
In Figure 3b the same system is used to demonstrate the specific lysis from expanded CD8+ve T cells sourced from the PBMCs of HLA-A2+ or HLA-A2-ve donors.
View Figure 3
These initial results suggest that manipulating the CD8-HLA class I interaction can have very significant effects on T cell activity and potentially on T cell specificity, which could both be of significant clinical utility.
Modifications of the CD8-HLA class I interaction could be potentially exploited in 3 ways as discussed below;
Until recently gene delivery mediated cancer therapy had struggled in clinical development. More recently clinical developments in cancer virotherapy have altered this with a licenced gene mediated therapy based on intratumoral injection [34]. With evolving technology now more ambitious gene mediated therapies that are activated only in designated targeted cancer cells seem a realistic future expectation [35].
The observation that target cells bearing the HLA-A2/Kb complexes can be killed by activated CD8 T cells of any HLA/peptide specificity suggests that delivery of these complexes by gene transfer could offer a new approach to therapy. The expectation is that expression of these complexes combined with activation of the immune system by either cytokines or PD1 inhibitors could lead to T cell mediated lysis of targeted tumour cells by activated normal T cells in all patients irrespective of their HLA type or pre-existing anti-tumour immune response.
Additionally, as the HLA-A2/Kb complexes produce activation and expansion of all T cells irrespective of native specificity, the presence of cells expressing A2/Kb complexes within the tumour environment may enhance the impact of PD1 based therapies on expanding and enhancing local tumour specific T cell responses against the largely unknown tumour epitopes [32].
With the new ability to effectively control T cell activation using drugs of low toxicity now established in routine clinical practice, the challenge now is how to selectively deliver and express the gene for HLA-A2/Kb or similar HLA derivatives with high affinity CD8 interactions.
This technology which closely parallels that of bispecific antibodies, has already had considerable development with a variety of systems using the standard HLA class I molecule. Whilst our recent work has, for ease of experiment, used transfected cells with cell surface protein expression, our previous work has indicated that antibody delivered HLA molecules have the same immunological recognition characteristics [36].
Recent publications from the team at Roche indicate that the technology of antibody-HLA targeting may be moving towards clinical applicability. However, the limited immunological specificity of the current targeting systems may well limit the efficacy and clinical utility. In the current formulations of the technology the targeting system includes only a single HLA/peptide complex. This will be recognised by virus specific T cells that are efficient cytotoxic CD8 T cells but by definition only comprise a very small proportion of the total CD8 T cell population. Additionally, there is the limitation that a range of differing constructs would be needed for patients of differing HLA tissue types.
The use of the HLA-A2/Kb molecule or related high affinity binding proteins could help overcome both of these issues as they will be recognised by the full range of T cells of all specificities and so harness cytotoxic CD8 T cell activity in each patient irrespective of patient HLA tissue type.
The use of an antibody delivery system to deliver the HLA-A2/Kb molecule may carry a risk that the HLA-A2/Kb molecule may be potentially immunogenic and could, unlike the standard HLA class I molecule, potentially lead to unwanted T cell activation distant from the tumour as circulating T cells interact with this new HLA construct. Forthcoming experiments examining the potential impact of soluble monomeric HLA-A2/Kb on T cell activation should provide insight into this risk.
The impact of the CD8-HLA class I interaction on determining both the degree of activation and specificity of the T cell target interaction is based on a manipulating a natural low affinity interaction.
The Table 1 below summarises the variation in the interactions, the affinity of the binding and the potential immunological outcomes.
Table 1: Summary of the impact of binding affinity changes in the CD8-Hla Class I interaction, the potential immunotherapy agonist and antagonist drug actions and their impact on immunological function. View Table 1
To date the affinity of this interaction ha+s been manipulated predominantly by using protein engineering approaches. A key question is, if using more modern technology, can the CD8-HLA class I interaction be either enhanced or weakened using drugs that target this protein-protein interaction? The development of drugs that inhibit or act as agonists in protein-protein interactions is now a rapidly evolving area of therapeutic research and development [37,38].
The 3D crystal structure of the CD8-HLA class I interaction is well characterised and some of the key amino acid interactions have been previously demonstrated. We have examined the crystal structure of the CD8-HLA class I interface to assess if it could be possible to inhibit or enhance this binding via novel drugs impacting on this protein-protein interface. A computer representation of the interface between these two molecules is shown below in Figure 4, including the area that may be suitable to be 'drugged'.
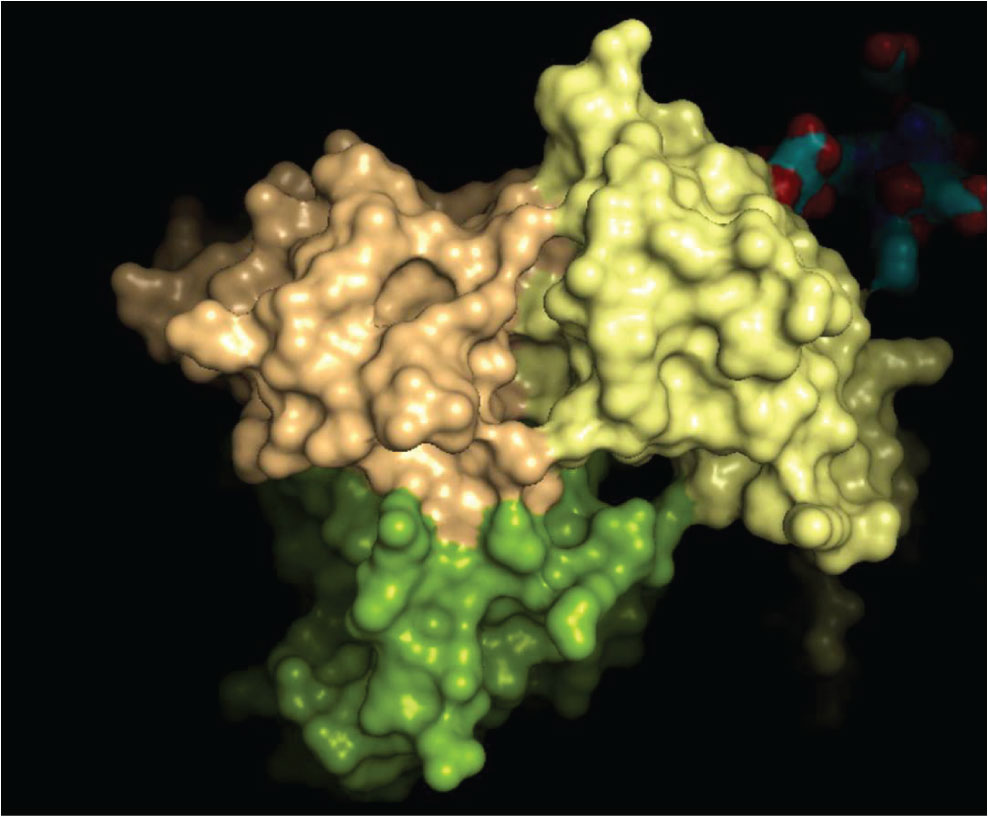 Figure 4: The HLA-CD8 complex (1AKJ crystal structure; HLA, yellow; CD8 domains in beige and green). The structures of the complexes show that there are several regions adjacent to contacts between chains that present cavities into which a small molecule could bind to exert agonist or antagonist properties.
View Figure 4
Figure 4: The HLA-CD8 complex (1AKJ crystal structure; HLA, yellow; CD8 domains in beige and green). The structures of the complexes show that there are several regions adjacent to contacts between chains that present cavities into which a small molecule could bind to exert agonist or antagonist properties.
View Figure 4
Previous work with peptides and recombinant soluble CD8 have shown significant immunological inhibition but have been too cumbersome to develop clinically [39,40]. More recent data using an antibody that binds to CD8 has demonstrated profound reductions in T cell activity [41]. Whilst the biological impact of this inhibition is important, the duration of activity with an antibody system may be too long for clinical application. Other technology that could achieve similar but more controllable inhibition may be of value in autoimmunity and the management of Checkpoint Inhibitor toxicity in cancer immunotherapy.
The protein engineering data indicates that increasing the affinity of the CD8-HLA class I interaction from a KD of 145 µM to 85 µM results in enhanced T cell activation and target cell killing whilst maintaining HLA/peptide specificity [32]. The key question is if a drug that moderately strengthens the HLA class I/CD8 protein-protein interaction will have a similar impact on enhancing T cell activation and killing abilities. If this is the case, then a drug with these characteristics that moderately increases the affinity of the CD8-HLA class I interaction may be of use an adjuvant to vaccination and also therapeutically as an approach to enhancing T cell responses to tumour cells in immune checkpoint inhibitor therapy.
A further increase in the affinity of the CD8-HLA class I interaction to a KD of 10 ºM using the HLA-A2/Kb- Supertarget leads to the loss of HLA/peptide specificity and this allows activated CD8 T cells of all specificities to kill targeted cells. The impact of this is demonstrated in Figures 3a and Figure 3b in which T cells are killing target cells bearing the HLA-A2/Kb molecule regardless of their normal HLA or peptide specificity.
In this situation, if a drug is able to strengthen the interaction it could lead to HLA and peptide independent killing by activated T cells in the environment where the drug is present. In this situation the ability of activated T cells to kill tumour cells would not be by the detection of specific tumour associated peptide, but by selective geographical targeting based on the targeted delivery of the strong agonist drug to the area of the tumour. A drug with this effect would have the potential to lead to there being T cell/T cell fratricidal killing as well as T cell/target cell killing. A therapy based on a strong agonist would clearly need the availability of a higher affinity inhibitory antagonist to be available to limit unwanted activity and toxicity.
This approach is clearly a novel concept and will require many key stopes in development however nanotechnology and antibody-based systems to selectively deliver other drugs/peptides selectively to tumours are already under development [42-44].
Tumour immunotherapy is now centre stage in cancer research and modern oncology practice, however despite the many advances the current technologies can be complex and often produce relatively modest results.
For a number of years, we and a number of other groups, have explored the use of antibody targeted HLA class I molecules to redirect T cells to kill tumour cells. However, the clinical utility of this technology may be limited due to the narrowness of the immune response exploited in the system. The recent data shown here indicates that the HLA-A2/Kb molecule can serve as a universal target to all activated CD8 T cells from any patient, suggests that efficacy and utility could be dramatically high with this revised system and a re-evaluation of the antibody-HLA system and parallel gene delivery systems is indicated.
Alongside this data, the modern developments in drug design suggests that it may be possible to manipulate the CD8-HLA class I interaction with novel drugs rather than protein engineering. It is possible that three simple drugs could be developed that would change this protein-protein. These drugs would allow the choice of down regulating T cell activity, moderately enhancing it, or non-specifically activating T cells which would allow T cells to kill targeted tumour cells irrespective of their native immunological identity.
At present for this application we have little data, however the basic science is supportive, the technology achievable and the potential rewards immense.
I would like to thank Professor Linda Wooldridge and Dr Anya Lissina for providing the data displayed in Figures 3a and Figure 3b. Thanks to Professor Scott Burrows for the use of the T cell clone SB10.