Purine Nucleoside Phosphorylase (PNP) deficiency in humans causes lymphopenia and this provided the rationale for developing PNP inhibitors as immunosuppressive agents. However, careful re-evaluation of clinical history of PNP deficient patients and clinical experience with PNP inhibitors together with new experimental data suggest inhibition of PNP may have immune activating effects through elevation of guanosine and activation of various toll-like receptors (TLRs). This paper proposes a mechanism of action for the immune activating effects of PNP inhibition.
To evaluate in vitro TLR activation, by PNP inhibitor and the purine nucleosides that are elevated with PNP inhibition, HEK293 cells expressing various TLRs linked to the SEAP reporter group was used. To evaluate forodesine as vaccine adjuvant, well known mouse models of tetanus toxoid vaccine and Hepatitis B vaccine was used. To evaluate anti-tumor and anti-bacterial effects of forodesine, syngeneic B16F10 mouse melanoma model and L.monocytogenes bacterial infection mouse model were used.
Guanosine demonstrated significant and robust activation of TLR2 and TLR4, in HEK293 cells. Like other TLR activators, PNP inhibitor forodesine which elevates guanosine in vivo demonstrated statistically significant increases in antibody titers and, although not significant, a trend towards increase in interferon-γ (IFN-γ) levels compared to vaccine alone groups in tetanus toxoid and Hepatitis B vaccine mouse models. Similarly, forodesine treatment improved the mouse immune system as demonstrated by significant decrease in tumor growth in mouse melanoma model, inspite of lack of direct in vitro cytotoxic effects of forodesine on malignant cells. In addition, like other TLR agonists, forodesine demonstrated significant decrease in weight loss in L.monocytogenes bacterial infection model.
In PNP inhibitor clinical trials, immune activating effects have been noted which included increased response to tetanus toxoid vaccine, graft versus leukemia effects in post hematopoietic stem cell transplant relapse acute lymphoblastic leukemia patients, and in vivo effectiveness in non-leukemic cancers in spite of lack of direct in vitro cytotoxic effects on the malignant cells. Clinical findings in PNP deficient patients include autoimmune manifestations, elevation of IL-18 levels (an IFN-γ inducer), and neurological disorders which is consistent with immune activation. Lymphopenia noted in PNP deficient patients is primarily due to constitutive activation of immune system in these patients causing immune exhaustion and T-cell elimination. Genetic studies, clinical experience with PNP inhibitors together with the preclinical data presented here clearly support the role of PNP inhibitors as immune-activating agents. Contrary to prior literature, we have confirmed that PNP inhibition has immune activating effects.
Clinical findings in PNP deficient patients along with clinical and preclinical experience with PNP inhibitor support the use of PNP inhibitors as an immuno-oncology agent and as a vaccine adjuvant.
Purine nucleoside phosphorylase, Forodesine, toll-like receptors, Vaccine adjuvant, Immuno-oncology agent
Purine nucleoside phosphorylase (PNP) catalyzes the breakdown of purine nucleosides, guanosine, deoxyguanosine, inosine and deoxyinosine, to the corresponding purine base and sugar 1-phosphate. Inhibition or deficiency of PNP in humans and mice results in elevation of these purine nucleosides in the serum and urine. PNP inhibition has historically been believed to lead to immune suppression. This is primarily based on observations from patients with genetic PNP deficiency which in all cases leads to lymphopenia [1,2]. Contrary to the observed immuno-compromised clinical phenotype of the PNP deficient subjects, we discovered that pharmacologic inhibition of PNP activates the immune system.
PNP-deficient children exhibit profound impairment in the T-cell component of their immune systems. This rare condition provided a model for the development of specific inhibitors of PNP as immunosuppressive agents for the treatment of autoimmune diseases and hematologic diseases [3-5]. Forodesine (NTR001) and ulodesine (NTR002) are two rationally designed, potent inhibitors of PNP that were advanced into the clinic [6,7]. Ulodesine was evaluated in patients with psoriasis and gout and, although safe, drug development was aborted [4,8]. Forodesine was advanced in hematologic malignancies and drug development was terminated in US and Europe although the drug did receive approval in Japan with limited data for the treatment of relapsed/refractory peripheral T-cell lymphoma [3,9-12]. In this paper, we will discuss how previous work on PNP inhibition/deficiency has overlooked its immune activating effects and discuss how these effects are manifested in PNP deficient patients and in patients treated with PNP inhibitors. Additionally, we will present pre-clinical data to elucidate the mechanism by which PNP inhibitors activate toll like receptors (TLRs) triggering immune activation and also explore possible clinical applications.
Purine nucleoside phosphorylase (PNP) deficiency is a disorder of the immune system (primary immunodeficiency) characterized by recurrent infections, neurologic symptoms, and autoimmune disorders. While infections usually begin in the first year of life, some patients remain asymptomatic until several years of age. The infections are similar to those found in severe combined immunodeficiency (SCID). They include sinusitis, pneumonia, urinary tract infections, pharyngitis, otitis, mastoiditis, thrush, and diarrhea. The autoimmune manifestation noted in this patient population include autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, rheumatoid arthritis, and systemic lupus erythematosus. Neurologic problems are common in PNP-deficient patients and these include spastic quadriparesis, spasticity, spastic diplegia, retardation of motor development, tremor, ataxia, hypertonia, hypotonia, developmental delay, hyperactivity, and mild to severe mental retardation [1,13].
PNP catalyzes the phosphorolysis of guanosine and deoxyguanosine, and inosine and deoxyinosine to guanine and hypoxanthine respectively. PNP deficient patients demonstrate high levels of PNP substrates guanosine, deoxyguanosine, inosine and deoxyinosine in the serum and urine. In addition, these patients show elevated levels of deoxyguanosine triphosphate (dGTP) in the erythrocytes [1]. The proposed mechanism for T-cell depletion is shown in Figure 1. Inhibition of PNP leads to elevation of deoxyguanosine which is phosphorylated to dGTP in lymphocytes. This occurs primarily in immature lymphocytes because of high levels of deoxycytidine kinase that can phosphorylate deoxyguanosine and low levels of nucleotidase that dephosphorylates deoxyguanosine nucleotides. Accumulation of dGTP results in imbalance of deoxynucleotide triphosphate (dNTP) pools, thereby causing misincorporation, DNA fragmentation and death of T-cells via a mechanism characteristic of apoptosis [14-17]. The high kinase and low nucleotidase levels are characteristics of immature T-cells - at normal PNP levels, there is no physiological consequence - however, when PNP activity is suppressed or absent, this aspect of human T-cells becomes significant as it allows for intracellular accumulation of dGTP.
Inhibition of PNP leads to immune activation through activation of TLRs (Figure 1). Inhibition or deficiency of PNP leads to elevation of guanosine and deoxyguanosine. We have recently discovered that guanosine activates TLR2 and TLR4 (data presented in preclinical section). In addition, guanosine can also activate TLR7 in the presence of RNA [18]. TLRs is one of the pattern recognition receptors (PRRs) that plays a key role in activation of innate immune system [19,20]. In response to microbe-specific molecular signatures known as pathogen-associated molecular patterns (PAMPs) and self-derived molecules derived from damaged cells, referred to as damage-associated molecular patterns (DAMPs), TLRs activate downstream myeloid differentiation response gene (MyD88) signaling pathway that leads to the induction of innate immune responses which include release of proinflammatory cytokines, maturation of dendritic cells, activation of NK cells (Figure 1). These processes are essential for the clearance of the insult as they not only trigger immediate host defensive innate immune responses but also prime and orchestrate antigen-specific adaptive immune responses.
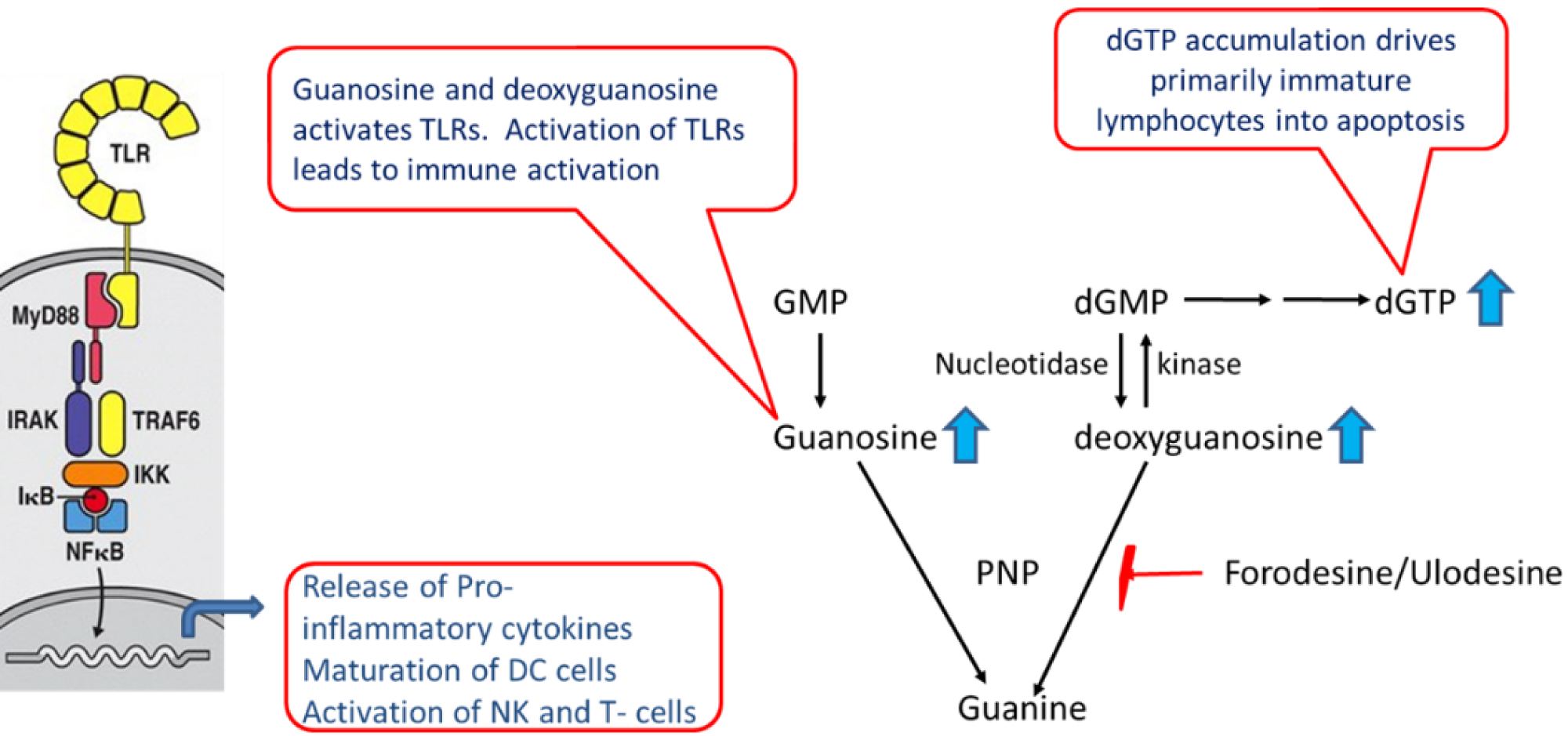 Figure 1: Role of PNP in immune modulation. Inhibition/deficiency of PNP leads to elevation of guanosine and deoxyguanosine that modulate the immune system through TLR activation and dGTP accumulation.
View Figure 1
Figure 1: Role of PNP in immune modulation. Inhibition/deficiency of PNP leads to elevation of guanosine and deoxyguanosine that modulate the immune system through TLR activation and dGTP accumulation.
View Figure 1
The role of TLRs in activation of the immune system is well known and currently a number of clinical trials are ongoing, investigating TLR agonists as immuno-oncology agents for the treatment of cancer as a single agent and in combination with check point modulators [21]. TLR agonist have also been approved as an adjuvant in the commercial vaccines [22,23]. However, much less appreciated is the concept that the innate immune system also utilizes these same TLRs to trigger immune suppression if continuously activated for the purpose of protecting the host from tissue damage. The phenomenon of T-cell immune-exhaustion/immune-suppression through persistent antigen exposure is well characterized in viral diseases [24]. Initial exposure to the viral antigen produces a robust immune response that can inhibit the viral replication as is noted with the primary immune response in patients newly infected with HIV [25]. However, chronic viral infections lead to constitutive activation of various PRRs which ultimately causes immune suppression through T-cell immune exhaustion and elimination resulting in disease progression. Similar observations have also been noted with certain bacterial and fungal infection [26,27].
Constitutive activation of TLRs leads to activation of various pathways that suppress the immune system and these include pyroptosis of immune cells, crosstalk with other PRRs like stimulator of interferon gene (STING), NALP3 and retinoic acid-inducible gene I (RIG-I; Figure 2), or modulating expression of various enzyme and cytokines. TLR ligands can induce pyroptosis, an inflammatory type of cell death caused by activation of NALP3 multiprotein complex known as an inflammasome complex. TLR agonists induce NF-κB-mediated NALP3 and pro-IL-1β expression, there by promoting NALP3 inflammasome assembly and caspase-1-mediated IL-1β and Interleukin-18 (IL-18) secretion and pyroptosis [28,29].
 Figure 2: Crosstalk between various PRRs leading to dynamic and opposing effects.
View Figure 2
Figure 2: Crosstalk between various PRRs leading to dynamic and opposing effects.
View Figure 2
Another pathway for TLRs to induce immune suppression is through the production of a metabolite called itaconate which activates anti-inflammatory transcription factor Nrf2 [30]. Itaconate also exerts anti-inflammatory effects by modulating macrophage metabolism and decreasing STING expression and responsiveness to STING agonists [31]. Additionally, activation of the innate immune response by TLR agonists is counteracted by the induction of immunosuppressive cytokines and enzymes including interleukin-10 (IL‐10), transforming growth factor‐β (TGF- β), indoleamine 2,3‐dioxygenase (IDO), and induced nitric oxide synthase (iNOS) [32].
When a TLR is constitutively activated each of these mechanisms act as a negative regulator counteracting activation of the immune system which then leads to immune exhaustion and eventual elimination of the T-cells. Genetic studies, clinical studies and preclinical studies discussed below with PNP deficiency/inhibitors support the concept that inhibition of PNP leads to TLR activation and constitutive TLR activation in PNP deficient patients maybe the cause of immune exhaustion and eventual elimination of T-cells.
Autoimmune manifestations, elevation of IFN-γ inducer IL-18 and neurologic disorders noted in PNP deficient patients can be rationalized with the concept of immune activation. PNP deficiency is a rare inherited disease characterized by profound T-cell immunodeficiency [1]. Infants with this disorder resemble SCID patients. In addition to recurrent infections, majority of these patients present with neurologic disorders and autoimmune manifestations. In some of the patients the neurologic symptoms become evident before the infections, suggesting neurologic disorders is not secondary to infection. The important thing to note here is that the clinical findings found more frequently in patients with PNP deficiency than in other patients with SCID are autoimmune disorders and neurologic problems.
So how do we reconcile these observations noted in PNP deficient patients with the concept of immune suppression or immune activation or both. Recurrent infection is consistent with the T-cell deficiency and immune suppression, fitting with the hypothesis that PNP inhibition may lead to immune suppression. Autoimmune diseases and neurologic disorders are generally characterized as related to hyperimmune activation and inflammation. Immune-suppressing agents are used to treat autoimmune diseases, making it difficult to rationalize autoimmune manifestations noted in PNP deficient patients with immune suppression. Neurologic disorders are generally associated with inflammation in the CNS compartment. Microglia, astrocytes and dendrocytes express high levels of TLRs especially in patients with neurologic disorders. Genetic studies and animal models suggest that TLRs do play a role in the inflammation of the CNS compartment [33,34]. Inhibition of PNP leads to activation of TLRs in the CNS compartment producing inflammatory cytokines and potentially causing neurologic disorders.
Recently a case report was published of a PNP deficient patient with a clinical presentation of systemic juvenile idiopathic arthritis onset complicated by macrophage activation syndrome [35]. In this PNP deficient patient serum CXCL9, a chemokine induced by interferon-γ (IFN-γ), was elevated. In addition, serum IL-18 level which is robust inducer of IFN-γ was also markedly elevated (> 400 fold). IL-18 was also measured in one more PNP deficient patient and found to be elevated to similar levels [36]. Activation of TLRs is known to cause release of IL-18 [37,38]. These observations further support the concept that inhibition of PNP leads to immune activation through TLR activation.
Constitutive activation of TLRs, through elevation of guanosine, in PNP deficient patient can also explain recurrent infection through immune exhaustion and elimination of T-cells. So, the question arises if the slow decline of T-cell function noted in PNP deficient patients is due to dGTP accumulation causing apoptosis in these cells or constitutive activation of T-cells through TLR activation leading to immune exhaustion and elimination of these cells or combination of both. The clinical findings in PNP deficient patients are consistent with the concept that PNP inhibition initially leads to immune activation, however PNP inhibition over a long period of time is somewhat similar to chronic exposure of antigen seen in viral infections, triggering immune exhaustion and T-cell elimination.
The two most advanced PNP inhibitors in the clinic are ulodesine and forodesine and the clinical experience with these two agents substantiate the role of PNP inhibitors as immune activating agent.
Ulodesine experience: Ulodesine was advanced into the clinic for the treatment of psoriasis and gout. Since it was believed that PNP inhibition causes immune suppression, the effect of ulodesine on response to tetanus toxoid vaccine was evaluated in gout patients [8]. The results were surprising as the percentage of patients responding to the vaccine were much higher in the ulodesine treatment arm compared to the placebo arm indicating that ulodesine acts as an adjuvant and enhances the immune response towards the vaccine.
Ulodesine was advanced in psoriasis patients as it is well known that T-cell immune suppressive agent can provide clinical benefit to these patients. No evidence of clinical efficacy was observed in any treatment arm in spite of decreases in T-lymphocytes in high dose group [4].
Forodesine experience: Forodesine was advanced into the clinic for the treatment of hematologic malignancies. Treatment of some of the post hematopoietic stem cell transplant (HSCT) relapse leukemia patients with forodesine led to facilitation of graft versus leukemia (GvL) effects, triggering donor lymphocyte expansion with clearance of leukemia, followed by GVHD which was managed by cyclosporin and steroids [10,38]. These patients did not develop significant GVHD at the time of HSCT but only showed GVHD during forodesine treatment. Unfortunately, GVHD and GvL effects are tightly linked as demonstrated by the inverse correlation between leukemia relapse rates and the severity of GVHD [39,40]. In a murine study of the MyD88, that mediates TLR signaling, recipients of MyD88 knock out grafts experienced unabated tumor growth. In contrast recipients of MyD88 wild type grafts showed evidence of GvL responses, suggesting an important role of TLRs on GvL [41,42]. Forodesine works by dual mechanism in leukemia patients. One is to increase dGTP in leukemia cells triggering cytotoxic effect in leukemia cells and the other is to facilitate GvL effect by donor lymphocyte activation and expansion through TLR and MyD88 signaling. The immuno therapies that have revolutionized cancer treatment, the check point modulators anti-PD1/PDL1 and anti-CTLA4, also demonstrated facilitation of GvL effects with increased GVHD, very similar to forodesine experience in this patient population [43].
Cutaneous T-cell lymphoma (CTCL) cells lines, in vitro, do not accumulate dGTP in the presence of PNP inhibitor and are not sensitive to PNP inhibition [44]. In spite of lack of direct cytotoxic effects on CTCL cell lines, in vitro, forodesine demonstrated clinical efficacy in CTCL patients [3,9,45]. Efficacy in CTCL patients can best be explained by immune enhancing effects of the PNP inhibitor.
Purine nucleosides and nucleotides were obtained from Sigma or MP Biomedicals. Cell lines and L.monocytogenes were obtained from ATCC and forodesine was bought from Aurum Pharmatech. Tetanus toxoid vaccine was purchased from Colorado serum company and ENERGIX-B™ was purchased from GlaxoSmithKline. Toll like receptor assays were performed at InvivoGen, San Diego CA. Hepatitis B vaccine study was performed at Institute of antiviral research, Utah State University, Logan, UT and the rest of the animal models were performed at Washington Biotechnology, Baltimore, MD. Animal studies were conducted in accordance with the approval of the Institutional Animal Care and was done in the AAALAC-accredited Laboratories.
TLR stimulation was tested by assessing NF-κB activation in HEK293 cells expressing a given TLR. The Secreted Embryonic Alkaline Phosphatase (SEAP) reporter is under the control of a promoter inducible by the transcription factor NF-κB in these cells. This reporter gene allows the monitoring of signaling through the TLR, based on the activation of NF-κB. In a 96-well plate containing the appropriate cells (50,000-75,000 cells/well), test articles purine nucleosides (100 µM) or forodesine (10 µM) or the positive control ligand is added. The media, DMEM containing Normocin™, added to the wells is designed for the detection of NF-κB induced SEAP expression. After a 16-24 hours incubation the optical density is read at 650 nm on a molecular device SpectraMax 340PC absorbance detector. This study was performed in triplicate.
Tetanus toxoid (TT) was used to vaccinate female swiss webster mice thrice, two weeks apart. Mice were treated by oral administration with forodesine and serum was collected at various time points for antibody titer and IFN-γ analysis. Mice were vaccinated subcutaneously with 0.1 ml tetanus toxoid on days 0, 14 and 28. Forodesine was administered orally one hour prior to vaccination. There were 6 mice per group. Sera from day 38 was used for antibody titers determination by ELISA using tetanus toxoid coated microtiter plates and anti-mouse conjugate. Sera from day 30 are assayed by ELISA for IFN-γ levels.
Female C57BL/6 mice were vaccinated IM with ENERGIX B™ on days 0, 21 and 43. A volume of 0.05 mL vaccine was injected IM in each leg for a total of 0.1 mL or 2 µg/mouse. There were 10 mice/group. Forodesine was administered intramuscularly (IM) or in the drinking water (DW). IM administrations of forodesine occurred on days of vaccination. Forodesine was administered in drinking water on days 0 through 57. Day 57 was the day of necropsy when serum and spleen was collected. Serum anti-HBsAg IgG was measured by ELISA. Splenocytes were suspended in RPMI medium, and adjusted to 5 × 106 cells per milliliter with growth medium. A volume of 2 mL of each splenocyte preparation was put into a well of a 12-well plate and stimulated with the recombinant HBs protein and incubated at 37 ℃ for 48 hours in a CO2 incubator. Splenocytes that were not stimulated with HBs protein was used as negative control. Supernatants were assayed using Mouse IFN-γ ELISA Kit.
B16F10 mouse melanoma cells (1 × 104 cells in 0.1 ml PBS with 20% Matrigel) were subcutaneously injected into the right flank of each female C57BL/6 mice. Administration of forodesine given by either oral gavage or drinking water was initiated on day 6 after injection of tumor cells. As a positive control one group was treated with cyclophosphamide, administered by IP. There were 10 mice/group. The animal weights and tumor volume measurements were recorded three times a week. Tumor volumes were calculated using the formula: Tumor Volume = length × width × width × ½.The animals were followed for 26 days post tumor cell injection.
Female Balb/c mice were injected intravenously with 0.2 ml L.monocytogenes suspension (1 × 106 CFU/mouse). Administration of forodesine was initiated 4 hours prior to the bacterial infection given either IP or PO or in DW. There were 10 mice/group. Weight loss and survival was monitored every day. The animals were followed for 10 days post L.monocytogenes infection.
Data are presented as means ± SEM and were analyzed with two-sided Student or Mann-Whitney test using GraphPad Prism software (Version 6.07, La Jolla, CA). Results were considered significant at P < 0.05.
Ulodesine and forodesine are potent inhibitors of PNP and, in the presence of deoxyguanosine, accumulate dGTP and inhibit proliferation of T-acute lymphoblastic leukemia (T-ALL) cell lines at low nM concentrations [6,7,17]. Proliferation of normal peripheral blood lymphocytes (PBLs), when activated with various antigens, are also inhibited in the presence of PNP inhibitor and deoxyguanosine [44,46]. However, PBL’s accumulate 10-fold lower dGTP levels and are also less sensitive to inhibition (EC50’s 4-20 fold higher) by PNP inhibitor and deoxyguanosine compared to T-ALL cells [17]. HUT102 cells, a T-cell line derived from a cutaneous T-cell lymphoma patient do not accumulate dGTP and proliferation is not inhibited in the presence of PNP inhibitor and deoxyguanosine [44]. The order of decreasing sensitivity to PNP inhibition in vitro through dGTP accumulation is as follows: T-ALL > PBLs > CTCL cells. The leukemic cells are highly sensitive to PNP inhibition as these are immature cells and known to have high kinase and low nucleotidase levels and that is needed for dGTP accumulation. On the other hand, lymphoma cells are mature T-cells and do not accumulate dGTP and are not sensitive to PNP inhibition.
To elucidate the mechanism of immune activation by PNP inhibitor, in vitro studies were performed in HEK293 cells expressing various TLRs in the presence of guanosine and other purine nucleosides that are elevated in vivo when PNP is inhibited. TLR agonist are known to act as a vaccine adjuvant, anti-tumor agent and anti-infective agent. So, PNP inhibitor was evaluated in mouse models of vaccines, melanoma and L.monocytogenes infection to further support the concept that inhibition of PNP leads to immune activation through TLR activation.
PNP deficiency/inhibition leads to accumulation of purine nucleosides, inosine, deoxyinosine, guanosine and deoxyguanosine, in the serum and urine [1]. Guanosine analogs like loxarabine and isotorabine are known to act as TLR7 agonist [47-49], so it was of interest to find out if any of the purine nucleosides that are elevated with PNP inhibition can activate TLRs. HEK293 cells transfected with various TLRs were used for evaluation of these purine nucleosides. The compounds were evaluated in triplicate and the results represent fold increase in activation of TLRs compared to the control group (no compound). Guanosine (100 µM) exhibited a significant stimulatory effect on human TLR2 and TLR4 (Figure 3). Inosine or PNP inhibitor, forodesine, had no effect on any of the TLRs. In addition, other purine nucleosides, deoxyinosine and deoxyguanosine, also showed no effect on TLR activation (data not shown). With inhibition of PNP, guanosine is elevated which can then act as TLR agonist and activate the innate immune system.
 Figure 3: Activation of TLR’s. Purine nucleosides (100 µM) and PNP inhibitor forodesine (10 µM) was assessed for TLR stimulation by assessing NF-κB activation in HEK293 cells, expressing a given TLR.
View Figure 3
Figure 3: Activation of TLR’s. Purine nucleosides (100 µM) and PNP inhibitor forodesine (10 µM) was assessed for TLR stimulation by assessing NF-κB activation in HEK293 cells, expressing a given TLR.
View Figure 3
PNP inhibitors ulodesine and forodesine in the mice inhibits PNP and increases purine nucleoside levels as seen in patients treated with these drugs [4,12,44,46]. Inhibition of PNP leads to elevation of guanosine in vivo and we have demonstrated that guanosine acts as TLR agonist. TLR agonists are used as adjuvant in commercial vaccines as they enhance the immune response to the antigen [50,51]. So, forodesine was evaluated in mouse tetanus toxoid vaccine model at two different doses administered by oral gavage, 30 mg/kg administered on the day of the vaccine and the following day and 60 mg/kg on the day of the vaccine only. All the groups received vaccine except the vehicle treated group. Tetanus toxoid vaccine when combined with forodesine significantly elevated the tetanus toxoid antibody titers in mice compared to the vaccine alone treated group (Figure 4A). Although not statistically significant, mice treated with forodesine (in the high dose group) potentially can increase Th1 immune response as shown by increase in serum IFN-γ levels (Figure 4B). Similar results were obtained with the use of another PNP inhibitor ulodesine in this model [52].
 Figure 4: Evaluation of antibody titers (4A) and IFN- γ levels (4B) in mouse tetanus toxoid vaccine model. Vaccinations was given on days 0, 14 and 28; Day 38 serum antibody titers and Day 30 serum IFN-glevels were measured. Forodesine - 30 +V (30 mg/kg/d given by oral gavage (PO) the day of vaccination and the next day) and forodesine - 60 + V (60 mg/kg/d given PO on day of vaccination only).
View Figure 4
Figure 4: Evaluation of antibody titers (4A) and IFN- γ levels (4B) in mouse tetanus toxoid vaccine model. Vaccinations was given on days 0, 14 and 28; Day 38 serum antibody titers and Day 30 serum IFN-glevels were measured. Forodesine - 30 +V (30 mg/kg/d given by oral gavage (PO) the day of vaccination and the next day) and forodesine - 60 + V (60 mg/kg/d given PO on day of vaccination only).
View Figure 4
Forodesine was also evaluated in HBV vaccine mouse model. All the groups received ENERGIX-B™ (HBV vaccine) vaccine except the vehicle treatment arm. ENERGIX-B™ in combination with forodesine, given in the DW every day at low dose of 2 mg/kg or given IM (60 mg/kg) on the day of vaccination only, stimulated antibody titers significantly greater than the titers from ENERGIX B™ vaccinated mice (Figure 5A). Forodesine in combination with ENERGIX-B™ treated groups demonstrated a trend towards increase in IFN-γ levels in supernatants of the splenocytes stimulated with hepatitis B surface antigen compared to the ENERGIX-B™-group, although not to a statistically significant level (Figure 5B).
 Figure 5: Female C57BL/6 mice were vaccinated with ENERGIX-B™ on days 0, 21 and 43. Forodesine was administered IM (Forodesine IM) at 60 mg/kg on days of vaccination. Another group was given forodesine at 2 mg/kg in DW (Forodesine DW) on days 0 through 57 the day of necropsy. Serum anti-HBsAg IgG end-point titers (5A) IFN-γ levels from supernatants of HBsAg-stimulated splenocytes (5B) were determined. Dotted line indicates values too high and out of the maximum range of the assay.
View Figure 5
Figure 5: Female C57BL/6 mice were vaccinated with ENERGIX-B™ on days 0, 21 and 43. Forodesine was administered IM (Forodesine IM) at 60 mg/kg on days of vaccination. Another group was given forodesine at 2 mg/kg in DW (Forodesine DW) on days 0 through 57 the day of necropsy. Serum anti-HBsAg IgG end-point titers (5A) IFN-γ levels from supernatants of HBsAg-stimulated splenocytes (5B) were determined. Dotted line indicates values too high and out of the maximum range of the assay.
View Figure 5
TLR agonists have demonstrated anti-tumor effects both in preclinical and clinical studies [53-55]. PNP inhibitor forodesine has no direct cytotoxicity effect on B16F10 mouse melanoma cells in vitro (data not shown). In the syngeneic mouse melanoma model, both forodesine treatment arms, 30 mg/kg given by PO every other week and 5 mg/kg given in DW every day, demonstrated significant decrease in tumor volume (Figure 6). In addition, in the low dose arm (5 mg/kg) 20% of the mice survived at the end of the study whereas in both the vehicle and high dose arm none of the mice survived. Cyclophosphamide was used as a positive control and, as expected, demonstrated significant decrease in tumor volume with 30% of the mice survived.
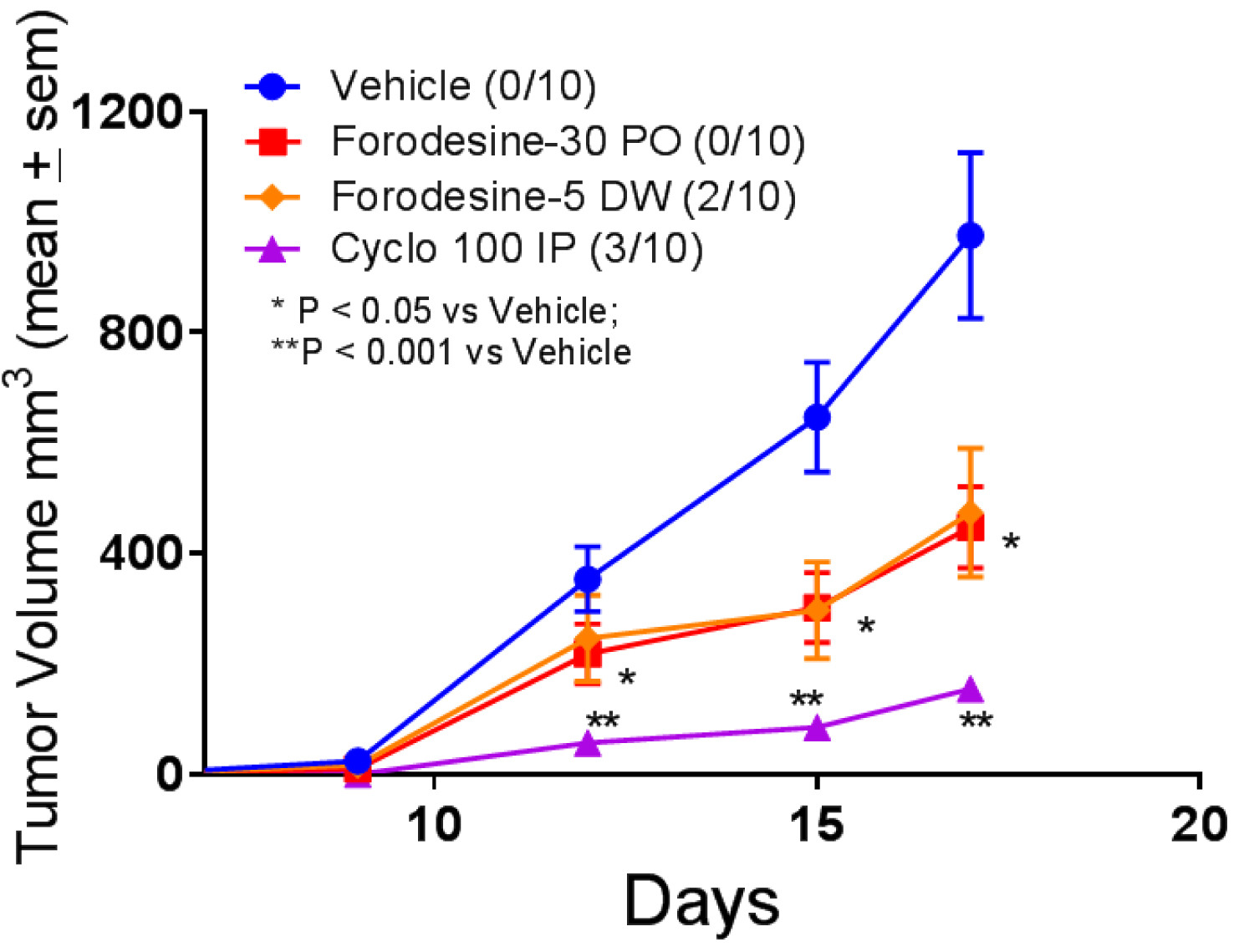 Figure 6: Evaluation of forodesine and cyclophosphamide (cyclo) in mouse melanoma model. B16F10 tumor cells were inoculated subcutaneously on day 0 and treatment was initiated on day 6. Forodesine was administered at 30 mg/kg PO (Forodesine-30 PO) starting day 6 every other week and the second group was administered 5 mg/kg in DW every day (Forodesine-5 DW) also starting day 6. Cyclophosphamide was given 100 mg/kg IP on day 6. The numbers in parenthesis indicate the number of animals survived.
View Figure 6
Figure 6: Evaluation of forodesine and cyclophosphamide (cyclo) in mouse melanoma model. B16F10 tumor cells were inoculated subcutaneously on day 0 and treatment was initiated on day 6. Forodesine was administered at 30 mg/kg PO (Forodesine-30 PO) starting day 6 every other week and the second group was administered 5 mg/kg in DW every day (Forodesine-5 DW) also starting day 6. Cyclophosphamide was given 100 mg/kg IP on day 6. The numbers in parenthesis indicate the number of animals survived.
View Figure 6
TLR agonists have also demonstrated efficacy in mouse models of bacterial and viral infection [49,56,57]. In mouse model of L.monocytogenes infection, all the forodesine treatment arms, 30 mg/kg IP and 30 mg/kg PO given on days 1-3 and 2 mg/kg in DW given every day, demonstrated significant decrease in weight loss (Figure 7). In addition, 10-20% of the animals survived in all the forodesine treatment arm whereas in the vehicle treatment arm none of the mice survived. The anti-bacterial effects noted in mouse model of L.monocytogenes infection further supports the concept of PNP inhibitors as immune activators. Similar results were obtained with the use of ulodesine in this model [52].
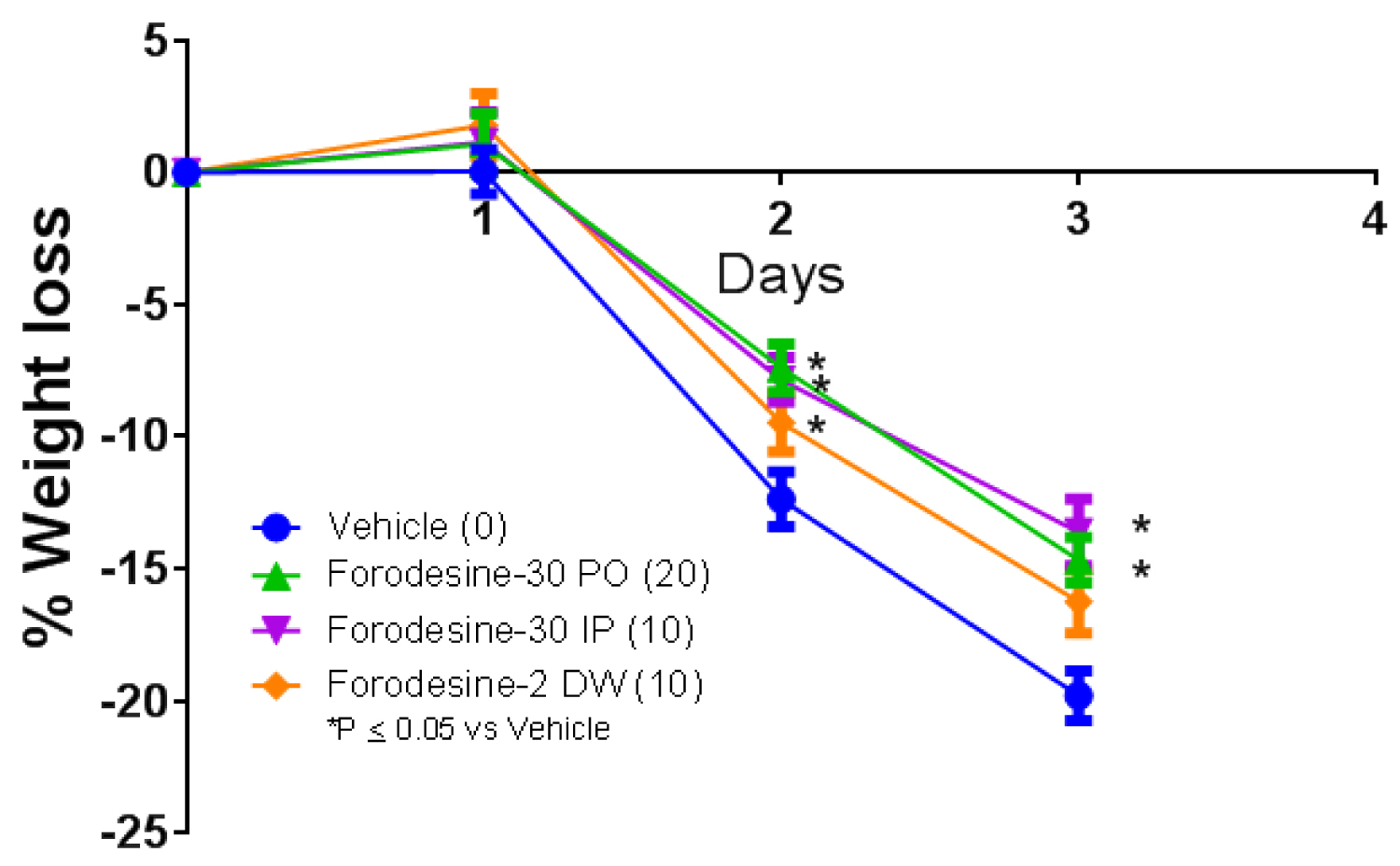 Figure 7: Bacterial (L. monocytogenes) suspension (1 × 106 CFU/mouse) was injected intravenously and the treatment was initiated 4 hours prior to infection. Forodesine was administered at 30 mg/kg IP or PO on days 1-3 (Forodesine-30 IP; Forodesine-30 PO). Another group was given forodesine at 2 mg/kg in DW every day till the end of the study (Forodesine-2 DW). Numbers in parenthesis indicate number of mice survived.
View Figure 7
Figure 7: Bacterial (L. monocytogenes) suspension (1 × 106 CFU/mouse) was injected intravenously and the treatment was initiated 4 hours prior to infection. Forodesine was administered at 30 mg/kg IP or PO on days 1-3 (Forodesine-30 IP; Forodesine-30 PO). Another group was given forodesine at 2 mg/kg in DW every day till the end of the study (Forodesine-2 DW). Numbers in parenthesis indicate number of mice survived.
View Figure 7
The historical belief that PNP inhibition leads to immune suppression may be true when there is PNP deficiency or PNP is inhibited completely and continuously for a long period of time. However pharmacologic inhibition of PNP actually activates the immune system through elevation of guanosine and activation of TLRs. We have demonstrated, using HEK293 cells expressing various TLR’s, that guanosine which is elevated with PNP inhibition activates TLR2 and TLR4. Shibata, et al. [18] demonstrated that guanosine and its natural analogs which include 8-OH guanosine, deoxyguanosine, and 8-OH deoxyguanosine activates TLR7 in the presence of RNA. Guanosine in the absence of RNA does not activate TLR7 which is consistent with our data and published literature [48].
TLR agonists are currently used as adjuvant in commercial vaccines to boost the immune response to the vaccine [50,51]. PNP inhibitor forodesine acts as a vaccine adjuvant and significantly increases the antibody titers in the mouse models of tetanus toxoid and Hepatitis B vaccine models. In addition, it also shows a trend towards increase in Th1 response by increasing the IFN-γ levels. These studies clearly indicate that PNP inhibitors acts as an immune activator. An immunosuppressive agent, like cyclosporin or mycophenolic acid, cannot act as an adjuvant and enhance the immune response to a vaccine. In addition, patients treated with ulodesine and vaccinated with tetanus toxoid also demonstrated increased immune response to the vaccine [8]. Boosting the vaccine response in preclinical mouse models and in the clinical studies unambiguously confirm the immune-activation potential of the PNP inhibitors.
TLR agonists have demonstrated anti-tumor effects both in preclinical and clinical studies [53-55]. Forodesine demonstrates anti-tumor effects in mouse melanoma model in spite of lack of direct in-vitro cytotoxic effects on the tumor cell line. In addition, clinical responses were noted in CTCL patients treated with forodesine inspite of lack of direct cytotoxic effects of forodesine on CTCL cell lines. The anti-tumor effects noted with forodesine in preclinical and clinical studies further support the role of PNP inhibitors as immune activators. TLR agonists have also demonstrated efficacy in mouse models of bacterial and viral infections [49,56,57]. Forodesine demonstrates anti-bacterial effects in L.monocytogenes mouse infection model which is again consistent with the concept that PNP inhibition leads to TLR activation and enhanced immune response.
Autoimmune manifestations are common and characteristic of PNP deficient patient [1]. In addition, significant increases in serum IL-18 levels, an IFN-γ inducer, seen in these patients is again consistent with the role of PNP inhibitors as immune activators [35-38]. Through elevation of guanosine, constitutive activation of TLRs in PNP deficient patient may be the cause of immune exhaustion and lymphopenia. The clinical findings in PNP deficient patients are consistent with the concept that PNP inhibition initially leads to immune activation; however, PNP inhibition over a long period of time is somewhat similar to chronic exposure of antigen seen in viral infections, causing immune exhaustion and T-cell elimination.
This new understanding of the role of PNP inhibitors in immune activation opens up multiple avenues for the use of PNP inhibitors in cancer treatment as an immuno-oncology agent or as an adjuvant in vaccine. It is important to emphasize that the dose and schedule of PNP inhibitor is going to be critical to use it as an immuno-oncology agent or as an adjuvant. Complete inhibition of PNP for a long period of time may be counterproductive as it can cause constitutive activation of TLRs causing immune exhaustion and T-cell elimination. Intermittent dosing or low dose given continuously may be more appropriate for immune activation by PNP inhibitors. STING agonist have been shown to potently induce antitumor activity in animal models of several cancers, including breast cancer, chronic lymphocytic leukemia, colon cancer, and squamous cell carcinoma. Recently, Sivick, et al. [58] demonstrated that STING agonists have differing anti-tumor effects at lower versus higher doses. In a mouse mammary tumor model, lower doses and/or infrequent dosing of STING agonist produced tumor specific CD8+ responses and provided better protection when re-challenged with the same tumor cells compared with mice treated with higher doses and more frequent dosing as mice in higher dose group showed limited to no immune response. Hence the dose and schedule of PNP inhibitor, or for that matter any immune activating agent, is going to be critical for its use as an immuno-oncology agent or as a vaccine adjuvant.
TLR therapy generally requires intra-tumoral injection or topical delivery because systemic delivery can promote non-specific immune activation, leading to safety and/or tolerability issues [53,59]. The major advantage of using PNP inhibitors as immuno-oncology agent is that the immune activation occurs in tumor micro-environment (TME) more so than in other tissues because of increased apoptosis and necrosis, due to hypoxia and high cell turnover in TME, causing an enormous increase in release of nucleic acids and nucleosides like adenosine and guanosine. Since PNP is an extremely efficient catalyst [60], guanosine is rapidly degraded in TME. However, in the presence of PNP inhibitor, guanosine remains elevated and activate the innate immune system through TLRs in TME. Although the level of guanosine has not been measured in TME, the level of adenosine has been measured and it is 10 to 100-fold higher in TME than in other tissues [61,62]. Because immune activation is more localized in TME there is less likelihood of immune related adverse effects commonly seen with other immunotherapies.
One additional advantage of PNP inhibitors is that the actual drug here is the metabolite guanosine (and its naturally occurring analogs) that is elevated with PNP inhibition. There is less potential for severe toxicity related to TLR activation because guanosine is limiting and once all the PNP is inhibited (which has been achieved in the clinic with the PNP inhibitors) higher doses of PNP inhibitors does not further increase the guanosine levels [4,11]. This explains the acceptable safety and tolerability profile of PNP inhibitors in the clinic with high doses and long-term exposure [3-5,8].
PNP inhibitors could be used in post stem cell transplant relapse patients to increase the activation and expansion of donor cells that could then control or eliminate the disease and restore donor chimerism. This phenomenon has been observed with forodesine in post stem cell transplant relapse leukemia patients [10,38]. PNP inhibitors could be combined with other immuno-oncology agents like CAR-T cells to increase the persistence of CAR-T cells in vivo through expansion and activation which may be beneficial in lymphoma patients. PNP inhibitors could also be combined with other immuno-oncology agents like checkpoint modulators (anti-PD1; anti-PDL1; or anti-CTLA4) and other anti-tumor agents which include chemotherapy, radiation and targeted therapies.
All the prior literature hypothesized PNP inhibitors as immunosuppressive agent. However, careful evaluation of the clinical findings of PNP deficient patients and surprising observations noted in patients treated with PNP inhibitors support the concept that pharmacologic inhibition of PNP leads to immune activation. Anti-tumor effects, anti-bacterial effects and robust increases in immune responses to vaccines in animal models presented here confirm the role of PNP inhibitors as immune activators. Thus, PNP inhibitors represent small molecule, orally bioavailable, novel, first in-class immune-activating agent, that can be used for the treatment of cancer and as vaccine adjuvant.
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Shanta Bantia- No conflict of interest.
NIAID funded the HBV vaccine study (Contract HHSN272201700041I, Task order HHSN27200010/A21). All the other studies were funded by Nitor therapeutics.
We acknowledge Dr. John Morrey and Neil Motter of Institute of Antiviral Research, Logan, UT for performing HBV vaccine model and providing valuable comments. We also acknowledge Dr. Nirmal Choradia of Palo Alto Veterans Affairs, Palo Alto, CA for his contribution in manuscript writing and for his valuable comments.