A novel, facile DNA detection method based on aggregation/disaggregation of an AIE (aggregation induced emission) dye-labeled peptide nucleic acid (PNA) probe was developed. This PNA probe plays the role of both target recognition and signal emission, thus it can quantify the target DNA without requiring additives such as intercalators and can distinguish the target from other sequences.
Aggregation induced emission (AIE), Tetraphenylethylene (TPE), Peptide nucleic acid (PNA), Telomere
In recent years, aggregation-induced emission (AIE) dyes have attracted attention as smart materials in the fields of luminescent materials [1], imaging [2] and sensors [3]. For sensors, many researchers have described applications for the detection of biomolecules such as ATP (adenosine triphosphate) [4], BSA (bovine serum albumin) [5] and G-quadruplex DNA using AIE dyes [6]. Generally, for these biomolecular detection methods, hydrophilic AIE dyes are used, which have hydrophilic parts such as a quaternary ammonium cation (R-N+R3) or a sulfonate anion (R-SO3-). In the cases mentioned above, electrostatic and/or hydrophobic interactions in aqueous media were used to recognize the target biomolecules. Thus, these methods require delicate and specific probe design for selective target detection. In contrast, if biomolecules are used to perform a specific recognition, highly selective detection can be attained. Therefore, we have studied sequence-specific detection of DNA by AIE-labeled peptide nucleic acids (PNAs), where the PNA contains the complementary sequence to the target DNA strands.
Because AIE dyes exhibit fluorescence-on switching in response to aggregation, the repetitive telomeric DNA [5'-(TTA GGG)-3'] is the target molecule of choice. The telomeric DNA terminal is elongated by telomerase in tumor cells, thus telomerase activity is an attractive candidate for early cancer diagnosis. Telomerase activity is generally estimated by the TRAP (Telomeric Repeat Amplification Protocol) assay [7-9], but it requires onerous methods such as gel electrophoresis. Other estimation methods of telomerase activity such as the HPA (Hybridization Protection Assay) [10], electrochemical assay [11,12], or ELISA (Enzyme-Linked Immunosorbent Assay) [13] also suffer from several problems. Recently, we have described a facile telomerase activity assay using a FRET (Fluorescence Resonance Energy Transfer) system constructed by a FAM (Carboxy Fluorescein Aminohexyl)-modified probe DNA and EB (ethidium bromide) as an intercalator [14]. This system can readily quantify the amount of telomeric DNA. However, EB is a suspected carcinogen [15], and an excess of EB was necessary with regard to the amounts of telomeric and probe DNA present.
Thus, we have synthesized an "AIE-labeled PNA complementary to telomeric repeat" system (AIE-Linker-CCC TAA-Linker-K) to construct a facile DNA detection method. PNA has the following attractive properties: i) High specificity to target DNA, ii) Extreme thermal stability of the duplex because of the absence of electrostatic repulsion. Hydrophobicity of the AIE dye enables aggregation of the AIE-PNA probe in aqueous media and consequently fluorescence may be induced (Signal-ON). Conversely, when AIE-PNA is hybridized with telomeric DNA, the AIE dye moiety will be dispersed in solution and fluorescence quenching will be induced (Signal-OFF). Therefore, the AIE-PNA probe is expected to work as an alternative method for the detection of telomeric DNA, without requiring an intercalator such as EB. In this paper, we describe the detection of telomeric DNA by fluorescence measurements based on the AIE-PNA probe method, where the AIE unit is a tetraphenylethylene (TPE) derivative (Figure 1), and investigate the operating principles of this DNA detection system.
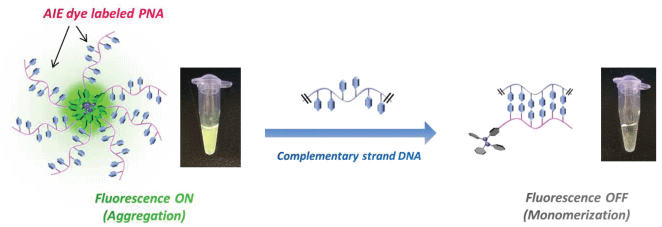 Figure 1: Surface modification of electrospun fibers and their applications. Radiation, wet or chemical treatments are used for the functionalization of nanofibers and different moieties can be tagged such as bioactive compounds or known drugs. Surface orientation of the nanofibers can be aligned or random and molecules can also be embedded within the fiber. Chemical or physical embedding can be done for functionalization. The functionalised fibres found promising applications in TE, 3D culture, drug delivery, wound healing and much more. View Figure 1
Figure 1: Surface modification of electrospun fibers and their applications. Radiation, wet or chemical treatments are used for the functionalization of nanofibers and different moieties can be tagged such as bioactive compounds or known drugs. Surface orientation of the nanofibers can be aligned or random and molecules can also be embedded within the fiber. Chemical or physical embedding can be done for functionalization. The functionalised fibres found promising applications in TE, 3D culture, drug delivery, wound healing and much more. View Figure 1
The AIE dye-labeled PNA probe was synthesized using the Fmoc peptide solid-phase synthesis method (SI). Synthesized and purchased DNA and PNA samples were dissolved in MilliQ water, and the concentrations of these solutions were determined from the absorbance at 260 nm. Measurement samples were prepared as follows: Telomeric DNA (12, 24, 48-mer) and negative control DNA (5'-TGA GTG TGA GTG-3') samples were diluted with MilliQ water (140 μL) and then 5 × tris-HCl buffer (100 mM Tris-HCl and 250 mM LiCl or KCl, pH 7.5, 40 μL) was added to each sample. The mixed solution was heated at 90°C for 2 min, and then cooled to room temperature. 30 μM of PNA probe (20 μL) was added to each sample (total volume 200 μL) and stirred with a vortex mixer. The expected fluorescence quenching was observed within seconds of addition of PNA probe to telomeric DNA. Actually, the fluorescence image shown in Figure 1 was taken at 5 min after the mixing of probe and DNA solutions. However, the fluorescent intensity of samples were varied widely especially in the case of blank or low concentrated samples when the measurement was performed just after the sample preparation. Thus, the fluorescence spectra of each sample were measured 12 h after the sample preparation.
Figure 2 shows the results of fluorescence measurements using synthetic telomeric DNA or negative control DNA. In the telomeric DNA samples, the fluorescence was quenched in proportion to the increase in telomeric DNA concentration. In the case of a high concentration of telomeric DNA (5 and 10 μM), the fluorescence of AIE dye was effectively fully quenched (quenching ratio > 90%). Meanwhile, negative control DNA did not hybridize with the PNA probe so the fluorescence intensity did not decrease. This result shows that the PNA probe alone is aggregated in aqueous solution, whereas in the duplex form, the AIE dye moiety is forced to disperse by the hydrophilicity of the DNA phosphate backbone. In other words, the phenyl rings of the AIE dye were permitted to rotate in the hybridized form, and this led to a non-radiative transition process. Consequently, fluorescence quenching occurred.
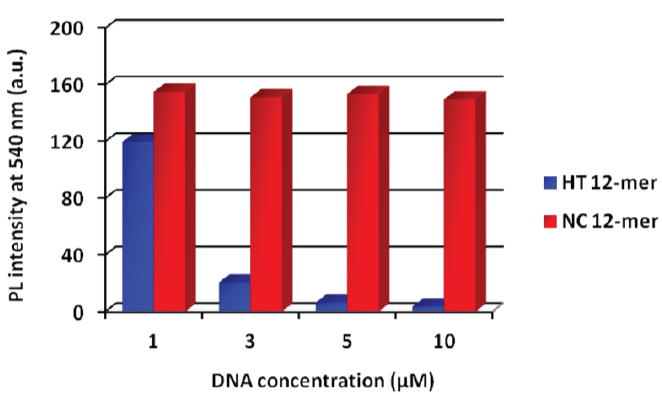 Figure 2: Fluorescence intensity at each DNA concentration. HT and NC represent human telomere and negative control, respectively. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMLiCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 2
Figure 2: Fluorescence intensity at each DNA concentration. HT and NC represent human telomere and negative control, respectively. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMLiCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 2
Figure 3 shows the results of fluorescence measurements in Li+ or K+ buffers. Telomeric DNA forms a quadruplex structure (G-quadruplex) in K+ buffer [16,17]. A G-quadruplex DNA cannot hybridize with the PNA probe, and we therefore expected that the fluorescence intensity of the PNA probe would not decrease. In the case of Li+ buffer, the fluorescence of the PNA probe was quenched according to the concentration of telomeric DNA 24-mer as well as telomeric DNA 12-mer. In contrast, no significant fluorescence quenching was observed in K+ buffer. Therefore, duplex formation was essential for the fluorescence quenching of the PNA probe and the G-quadruplex form did not affect the fluorescence intensity. These results indicate that this method was applicable to detecting specific hybridization between the PNA probe and the target DNA. We then carried out a quantitative analysis of the amount of telomeric DNA using this detection system (Figure 4). Because the actual telomeric DNA is quite long, a 48-mer telomeric DNA was chosen as the model sequence. The fluorescence intensity at 540 nm linearly decreased with increasing telomeric DNA concentration over the range of 0-1 μM. At a higher telomeric DNA concentration range (> 1 μM), fluorescence intensity was only slightly further reduced. This means that the linear dynamic range of this detection system was 0-1 μM of telomeric DNA under the present conditions. Interestingly, the fluorescence quenching ratio was decreased by the use of the longer telomeric DNA as the target molecule (refer to the supplementary Figure S2 (Supplementary file)). If each probe became bound to an individual target DNA strand as shown in the bottom image of Figure 5, then in the presence of an excess amount of target DNA, the initial probe aggregates should become fully dispersed in the solution according to the hydrophilicity of remaining (overhang) single strand and essentially complete fluorescence quenching should be expected. However, in fact, the smaller fluorescence quenching was observed in the case of longer telomeric DNA (refer to the supplementary Figure S2 (Supplementary file)).
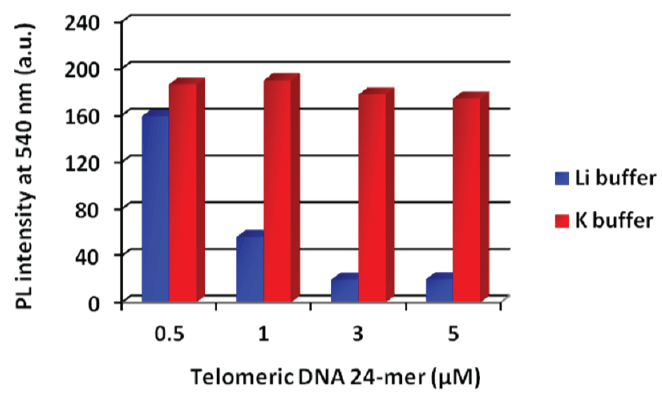 Figure 3: Fluorescence intensity at each DNA concentration in each buffer solution. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMeach salt (Li+: LiCl, K+: KCl) and 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 3
Figure 3: Fluorescence intensity at each DNA concentration in each buffer solution. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMeach salt (Li+: LiCl, K+: KCl) and 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 3
According to these results, it is conceivable that an "allosteric-like" binding occurred between the longer target DNA and the PNA probes and that re-aggregation occurred because of the hydrophobicity of multiple AIE dye molecules bound to individual DNA strands (Figure 5). The probes exist as aggregates, thus it is reasonable to believe that multiple probes would bind preferentially to single DNA strands rather than via one-to-one binding. In the case of previously reported DNA sensing by AIE-labeled DNA [18], fluorescence was turned on by duplex formation, and they concluded the restriction of molecular motion was the main reason for the change in fluorescence. However, in the present method of DNA sensing by the AIE-PNA probe, fluorescence quenching was mainly caused by dispersion of the AIE dye moiety. It was supported by the fluorescent measurement in mixed solvent of acetonitrile and water. The fluorescence observed in water was quenched in 50% acetonitrile/water solution.
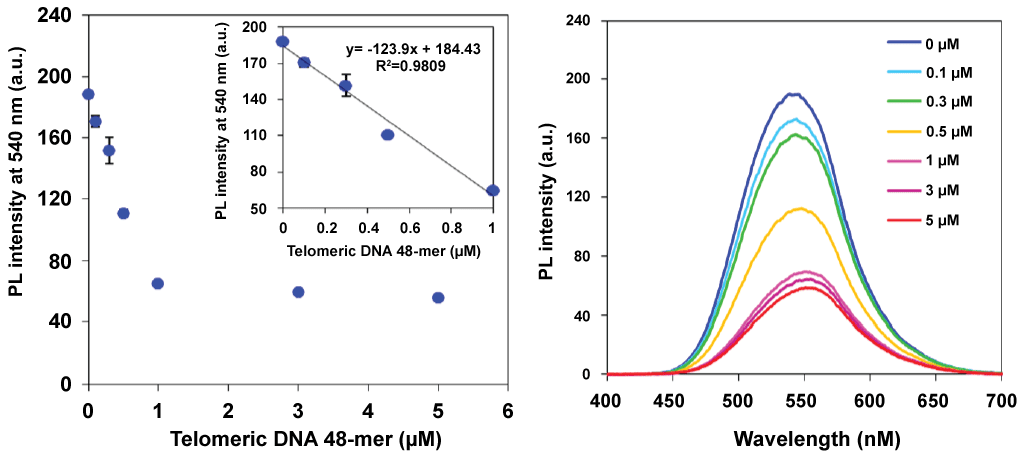 Figure 4: Relationship between the fluorescence intensity and the concentration of the telomericDNA 48-mer with enlarged inset (left), and fluorescence spectra of the PNA probe with the telomericDNA 48-mer (right). Error bars represent SD from n = 3. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMLiCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 4
Figure 4: Relationship between the fluorescence intensity and the concentration of the telomericDNA 48-mer with enlarged inset (left), and fluorescence spectra of the PNA probe with the telomericDNA 48-mer (right). Error bars represent SD from n = 3. Each sample was excited at 360 nm. [PNA probe] = 3 mM, 50 mMLiCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 4
 Figure 5: Illustration of the binding mode of the PNA probe with short-chain (top) and long-chain (center, bottom) telomericDNA. View Figure 5
Figure 5: Illustration of the binding mode of the PNA probe with short-chain (top) and long-chain (center, bottom) telomericDNA. View Figure 5
We performed DLS (Dynamic Light Scattering) measurements to confirm the initial formation of PNA probe aggregates. The PNA probe concentration for DLS measurements was 10 μM because a 3-μM solution was too dilute. Figure 6a shows the size distribution of PNA probe aggregates in the buffer solution. The average aggregate size was about 10 nm. These aggregates may form nanoparticles such that the hydrophobic AIE dyes are located inside the aggregates, and thus they can strongly emit fluorescence because of the restriction of intramolecular rotation.
In contrast, when duplexes were formed between the PNA probe and telomeric DNA, almost no 10-nm size particles were observed, i.e. the probe aggregates were disrupted by the hybridization and dispersed as smaller duplexes (Figure 6b).Consequently, the phenyl rings in AIE dyes were permitted to rotate, and the fluorescence was weakened by the increase in the non-radiative transition process of the duplex structures. Figure 6c shows the size distribution in K+ buffer, in which 24-mer telomeric DNA formed a G-quadruplex structure. This DLS spectrum contains the contribution from a large amount of small G-quadruplex particles. Nevertheless, a shoulder peak arising from probe aggregates can be observed. This shoulder arose because the PNA probes could not hybridize with the G-quadruplex DNA, and probe aggregates thus survived as fluorescent nanoparticles. The fluorescence quenching of AIE-PNA probes was ascribed to the collapse of fluorescent aggregates through duplex formation between the probe and target DNA, and therefore this method is applicable to the detection of specific DNA targets.
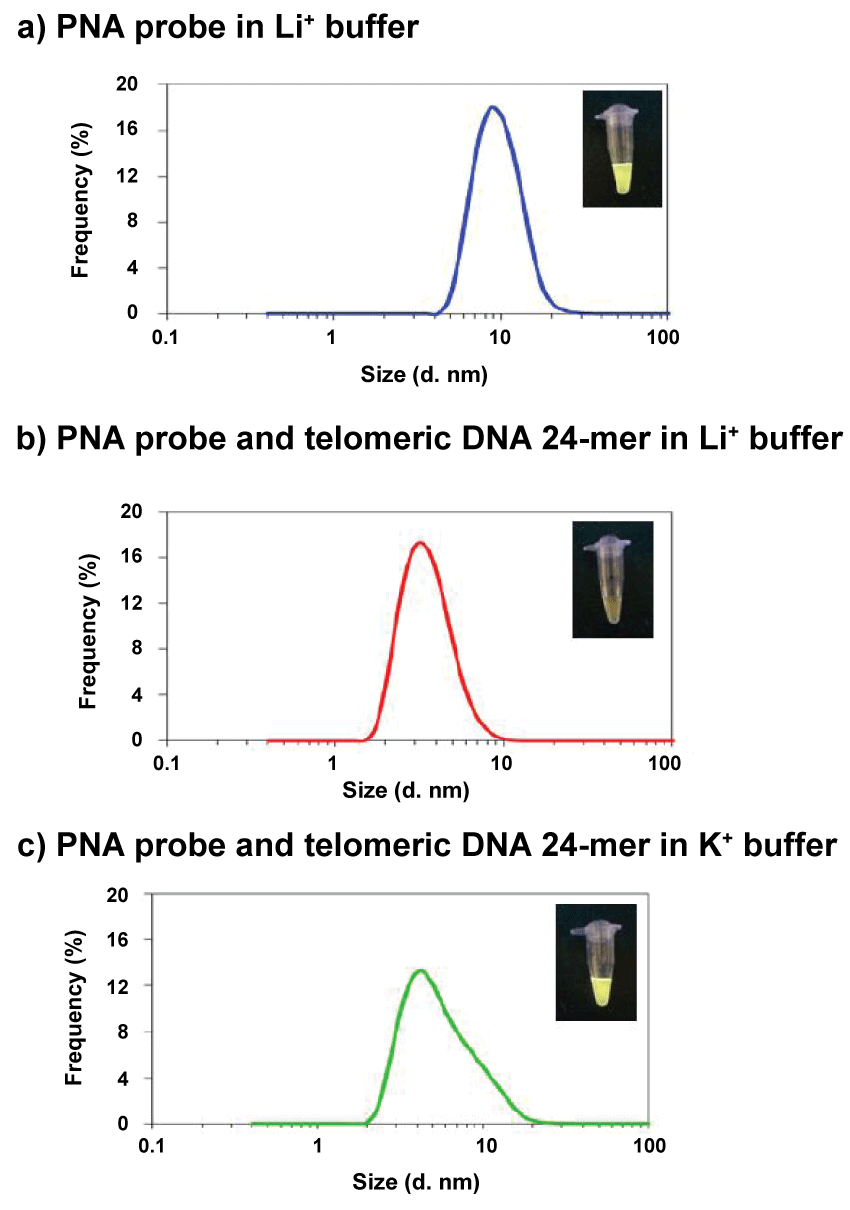 Figure 6: Relationships between the particle size and frequency under each condition, (a) PNA probe only in Li+buffer, (b) PNA probe and telomericDNA 24-mer in Li+buffer, (c) PNA probe and telomericDNA in K+buffer. [PNA probe] = 10 mM, [telomericDNA] = 20 mM, in 50 mMLiClor KCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 6
Figure 6: Relationships between the particle size and frequency under each condition, (a) PNA probe only in Li+buffer, (b) PNA probe and telomericDNA 24-mer in Li+buffer, (c) PNA probe and telomericDNA in K+buffer. [PNA probe] = 10 mM, [telomericDNA] = 20 mM, in 50 mMLiClor KCland 20 mMtris-HClbuffer (pH 7.5), 25°C. View Figure 6
We constructed a facile DNA detection system using AIE dye-labeled PNA probes based on fluorescence-switching by dye aggregation/disaggregation, which triggered extreme changes in the fluorescent signal. This PNA probe can quantify target DNA without requiring additives such as an intercalator and it can distinguish the target from other sequences or other structures derived from the same target sequences. Although the fluorescence quenching ratio decreased with the use of a long telomere DNA, the fluorescence intensity varied inversely in proportion to the concentration of the target, and was still sufficient for detection. Thus, this detection system can be applied for repetitive long-chain target DNA. This novel DNA detection system does not require the use of two fluorescent dyes unlike the Molecular Beacon, FRET and iFRET (induced FRET) methods. In addition, this detection technique will enable multiple DNA target detection within a single system because it will support detection via a specific dye for each sequence.
This work was supported in part by Grants-in-Aid for Scientific Research (C) (grant number 25460708) from the Japan Society for the Promotion of Science (JSPS).