Charcot-Marie-Tooth disease (CMT) is a hereditary sensorimotor polyneuropathy which is occasionally associated with demyelinating lesions in the central nervous system, although it remains unclear whether this finding is coincidental or whether the two processes share a common pathogenic mechanism.
A male patient with sensorimotor polyneuropathy presented with symptoms consistent with cauda equina syndrome at age 47 in relation to an inflammatory lesion in the conus medullaris. Six years later, he presented with sudden-onset neurological symptoms including dysarthria and right hemiparesis. Brain MRI revealed multiple inflammatory/demyelinating lesions. In both episodes, the CSF study showed mild hyperproteinorrachia without pleocytosis or oligoclonal IgG bands (OCBs), and the patient showed good response to corticosteroids. Genetic analysis revealed a duplication of region 17p11.2 of the PMP22 gene, typical of CMT1A.
Current clinical and experimental data appear to support an association between demyelinating diseases of the peripheral and central nervous systems based on the expression of common proteins that may function as target antigens and therefore direct the immune response against both neurological systems. This appears to be the case in CMT1A in which PMP22 is expressed in peripheral nerve myelin, in the brain and in the spinal cord.
Polyneuropathy Charcot-Marie-Tooth disease, PMP22, Demyelinating disease, Myelin, Oligoclonal bands
Charcot-Marie-Tooth disease (CMT) is a hereditary motor and sensory polyneuropathy which can be either demyelinating (Type 1) or axonal (Type 2), with an autosomal dominant, autosomal recessive or X-linked inheritance pattern [1]. Mutations in the MFN2, GJB1, MPZ, NDRG and PMP22 genes have been associated with demyelinating central nervous system (CNS) lesions [2-6]. Mutations in the 17p11.2 region of the peripheral myelin protein 22 gene (PMP22) cause CMT1A when the region is duplicated, and hereditary neuropathy with liability to pressure palsy (HNPP) when it is deleted. Both genotypes are associated with demyelination of the CNS [6,7]. In these cases, the clinical presentation may mimic multiple sclerosis (MS) or ischaemic cerebrovascular disease. The combination of central and peripheral involvement in CMT is of great clinical relevance, since it is necessary to establish a differential diagnosis with a primary demyelinating CNS disease such as MS, mainly due to the different prognosis and the iatrogenicity resulting from inappropriate treatment choices.
We present the case of a patient with recurrent neurological clinical manifestations consistent with demyelinating CNS disease in the context of CMT1A.
A male patient who was admitted to the hospital at the age of 47 years due to symptoms consistent with cauda equina syndrome (lower limb weakness and numbness, perineal hypoesthesia and bladder dysfunction); the examination performed on admission revealed abnormally high arches of the feet (pes cavus) and universal areflexia. Spinal magnetic resonance imaging (MRI) revealed the presence of a slightly contrast enhancing spinal cord lesion. The CSF study showed an absence of cells and IgG OCBs, and hyperproteinorrachia (67 mg/dL). Treatment was initiated with dexamethasone, which led to resolution of the symptoms. The follow-up lumbosacral MRI scan was reported as normal.
At 53 years of age, the patient was readmitted to our unit due to sudden-onset symptoms consisted ofdysarthria and right hemiparesis. Brain MRI was performed which showed several T2/FLAIR hyperintensities and notably the presence of a contrast-enhancing pseudonodular lesion in the left corona radiata causing diffusion restriction, prompting us to consider the possibility of inflammatory-demyelinating CNS disease in the differential diagnosis. Tests to rule out ischaemic cerebrovascular disease were negative. A new CSF study was carried out which continued to show hyperproteinorrachia (62 mg/dL) without pleocytosis and with no oligoclonal IgG bands. Neurophysiological study of visual evoked potentials (VEP) identified a discrete and symmetrical prolongation of P100 latency (117 ms). It was decided to start treatment with bolus of 1 g of 6-methylprednisolone, which led to a clear improvement in neurological deficit.
After a 13-year absence, the patient was referred to our unit by his primary care physician for investigation of his polyneuropathy. There had been no new clinical recurrences to date and the phenotype was compatible with CMT hereditary polyneuropathy, with universal areflexia, muscle atrophy in the legs, pes cavus, weakness of ankle dorsiflexion and bilateral steppage gait. Electroneuromyography showed signs of severe sensorimotor polyneuropathy: 1) Sensory nerve action potentials were not detected inmedianus, radialis, ulnaris and suralis nerves, 2) Compound muscle action potentials were not detected in peroneus profundus and tibialis nerves and severely reduced in medianus and ulnaris nerves, 3) Motor conduction velocities were severely decreased in medianus and ulnaris nerves, and 4) Denervation potentials were detected in cuadriceps, tibialis anterior and gastrocnemius muscleson needle electromyography study. A sural nerve biopsy was performed to rule out a systemic inflammatory or infiltrative process, showing severe loss of myelinated nerve fibres (Figure 1), and a new brain MRI scan showing the lesions reported during his second hospital stay, but no new lesions or contrast uptake (Figure 2). A genetic study was ordered which revealed duplication of the 17p11.2 region which encodes the PMP22 protein responsible for CMT1A.
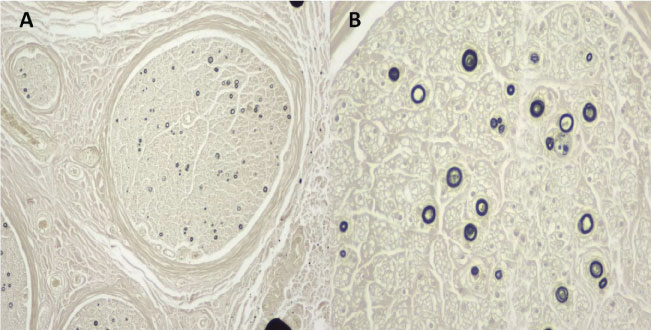 Figure 1: A) (Osmium 4X): Nerve fascicles showing significant loss of more than 90% of myelinated nerve fibres of all diameters, sparing only isolated fibres; B) (Osmium 10X): Spared fibres showing thinning of the myelin sheath. No axonal regeneration groups observed. View Figure 1
Figure 1: A) (Osmium 4X): Nerve fascicles showing significant loss of more than 90% of myelinated nerve fibres of all diameters, sparing only isolated fibres; B) (Osmium 10X): Spared fibres showing thinning of the myelin sheath. No axonal regeneration groups observed. View Figure 1
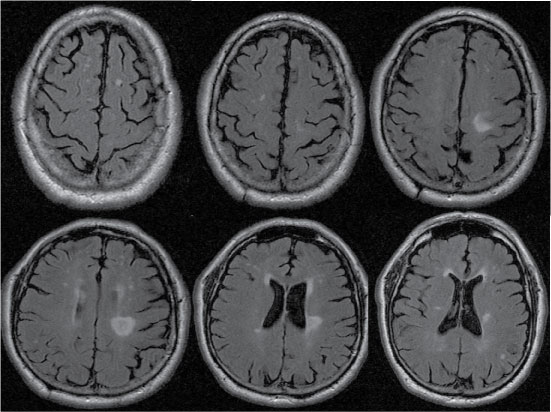 Figure 2: Brain MRI with FLAIR sequence. Multiple hyperintense lesions in the brain parenchyma, and one pseudonodular lesion in the left corona radiata. View Figure 2
Figure 2: Brain MRI with FLAIR sequence. Multiple hyperintense lesions in the brain parenchyma, and one pseudonodular lesion in the left corona radiata. View Figure 2
While the presence of inflammatory-demyelinating CNS lesions in peripheral neuropathies is a controversial topic, this association is well established in the case of chronic inflammatory demyelinating polyneuropathy (CIDP) [7,8]. The concurrent presence of central and peripheral demyelination prompts us to consider whether this is a single entity with common pathogenic mechanisms, or a mere coincidence of two pathologies. This combination is clinically heterogeneous and usually triggered by an infectious event. Its onset can be central or peripheral, with a monophasic or recurrent course, poor response to treatment with corticosteroids and immunoglobulins, and a prognosis which is generally poor [9]. Although the pathogenesis has not been fully elucidated, in the context of CIDP, it has been linked to the presence of anti-neurofascin 155 antibody, and it would seem that in this case, it could constitute an entity of its own with specific clinical characteristics [10]. Several studies have shown a very high incidence of sensorimotor neuropathy in patients with multiple sclerosis [11,12]. However, peripheral nerve disorders in neuromyelitis optica (NMO) have been associated with radiculitis, and aquaporin-4 deposition at the transition between the central and peripheral nervous systems at the spinal root level has been established as a possible pathogenic cause [13].
Although MS and CMT are classified as minority diseases based on their incidence, they are the most prevalent entities within their nosological groups and could feasibly coincide in one same individual. There are case reports in the scientific literature that clearly suggest the concomitance of both diseases [14], although others point towards CNS manifestations of a primary process, genetically determined by a single nucleotide polymorphism (SNP) [15]. In this regard, it has been suggested that MS and CIDP have different CSF inflammatory marker profiles [16], and cases of CMT with brain lesions are not usually associated with the presence of oligoclonal IgG bands in the CSF [4-6,15,17], which suggests that the two clinical entities differ in their pathogenesis.
Cases of CNS involvement have been reported in both subtypes of CMT and with different genetic mutations, e.g. in CMT1A (PMP22), CMT2A (mitofuscin2) and X-linked CMT (CMTX) due to mutations in GBJ1. CMT1A is caused by a duplication of the 17p11.2 region of the PMP22 gene, while deletion of this region gives rise to HNPP, and both polyneuropathies are associated with CNS abnormalities which can be asymptomatic in some cases and associated with neurological (e.g. pyramidal) symptoms in others. There appears to be a causal link with this kind of CNS involvement; in this regard, a case of familial CMT1A involving a father and daughter was reported (with pyramidal signs in the father and hyperintense lesions on T2-weighted magnetic resonance imaging in the daughter) [17] and one family with NHPP, in which affected relatives from two generations showed hyperintense CNS lesions, while asymptomatic patients had a normal brain MRI [18].
A possible physiopathogenic explanation that would interrelate concomitant inflammatory-demyelinating disease in the central and peripheral nervous systems in the case of different types of CMT may lie in the detection of the PMP22 protein, mitofuscin2 and GBJ1 in different structures of the CNS (optic nerves, brain and spinal cord), which could trigger an autoimmune response against these proteins [19,20]. An alternative hypothesis could be that autoimmunity is induced via molecular mimicry, as PMP22 shares partial homology with other CNS proteins like the proteolipid protein [6,14]. The presence of these proteins in these different locations of the CNS would explain the clinical findings (myelitis and encephalitis) and subclinical visual pathway involvement in our patient. The excellent clinical response to corticosteroids also appears to speak in favour of inflammatory-demyelinating disease of the CNS, while the repeated absence of oligoclonal IgG bands (intrathecal synthesis) in CSF would suggest that this is not an inflammatory disease of the CNS that fits the profile of multiple sclerosis.
We reported a case of CMT1A in a patient who presented with recurrent cerebral-spinal neurological symptoms in whom it was hypothesized that the CNS lesions, which exhibited an inflammatory-demyelinating profile, shared a common pathogenesis with demyelinating peripheral neuropathy. The response to corticosteroids was excellent, with a clinical prognosis conditioned by the polyneuropathy rather than CNS involvement. The absence of oligoclonal IgG bands in the CSF of our patient was crucial to the proper characterization the clinical entity.