To analyze the peripheral pulmonary vessels in patients with pulmonary hypertension (PH) on consecutive HRCT without slice gap and 3D-VR (volume rendering)-CT.
We studied 40 (17-82 mean 45.6-years-old, F/M = 24/16) patients with clinically verified pulmonary hypertension, reviewing the pulmonary angiography, HRCT and 3D-CT. This study includes both idiopathic PAH (n = 20) and secondary PAH (n = 20). The causes were as follows; CTEPH (chronic thrombo-embolic PH) (n = 7), collagen vascular diseases (MCTD1, SjS1, RA1, SLE1), ASD (n = 2), VSD (n = 2), aortic coarctation with PDA (n = 1), liver cirrhosis (n = 1), mitral valve regurgitation (n = 1), single atrium and ventricle (n = 1), and Rendu-Osler-Weber disease (n = 1).
The prominence of the pulmonary vasculature and/or tortuosity (n = 16) and dilatation of the central PA (n = 39) with RV hypertrophy were frequently observed. The interlobular septal thickening (n = 18, 45%), and centrilobular (perivascular) ground glass opacities (n = 6, 15%) were clearly depicted on gapless HRCT. PA aneurysmal dilatation and irregular dilatation with arterio-venous fistula were observed in 2 (5%) and 3 cases (7.5%), respectively. Bronchial artery dilatation was observed in 5 cases. RV myocardial muscle hypertrophy was observed in 4 cases (10%). The lymphadenopathy caused mainly by edematous reaction was observed in 9 (22.5%) cases.
Besides prominence of the pulmonary vasculature and/or tortuosity of PA, the centrilobular (perivascular) ground glass opacities (n = 6, 15%) reflecting perivascular edema and/or hemorrhage were detected on HRCT. PA aneurysmal dilatation and irregular dilatation with arterio-venous fistula were observed in 2 (5%) and 3 cases (7.5%), respectively in severe PH patients.
Pulmonary hypertension, Pulmonary arteriovenous fistula, Pulmonary artery aneurysm
AO: Aorta; ASD: Arterial Septum Defect; AVF: Arteriovenous Fistula; BA: Bronchial Artery; BP: Blood Pressure; COPD: Chronic Obstructive Pulmonary Disease; CPFE: Combined Pulmonary Fibrosis and Emphysema; CTR: Cardiothoracic Ratio; GGO: Ground Glass Opacity; ILS: Interlobular Septum; IP: Interstitial Pneumonia; I-& S-PH: Idiopathic & Secondary-Pulmonary Hypertension; LA: Left Atrium; LN: Lymphadenopathy; MCTD: Mixed Connective Tissue Disease; MPR: Multiple Planner Reconstruction; PA: Pulmonary Artery; PAP: Pulmonary Arterial Pressure; PAR: Pulmonary/Arterio Ratio; PV: Pulmonary Vein; PDA: Patent Ductus Arteriosus; RA & RV: Right Atrium & Ventricle; RA: Rheumatoid Arthritis; SLE: Systemic Lupus Erythematosus; Sjs: Sjogren's Syndrome; VSD: Ventricle Septum Defect
1. The prominence of the pulmonary vasculature and/or tortuosity and dilatation of the central pulmonary arteries were frequently observed in severe PH patients.
2. The peripheral PA aneurysm and irregular dilatation of PA with PV fistula were revealed in two (5%) and 3 cases (7.5%) on gapless HRCT.
CT is capable of estimating the etiology and pathophysiology of pulmonary artery hypertension (PH), and can be useful in the management of these patients. Pulmonary artery and arterioles are nearly always affected, irrespective of its cause. Here, we report and discuss the new imaging findings of consecutive HRCT without slice gap, MPR reconstructed views and 3D-VRCT including contrast enhancement study. The PH is the hemodynamic consequence of vascular changes within the pre-or post-capillary pulmonary circulation. The PH is hemodynamically defined as a condition in which the mean pulmonary artery pressure (PAP) is greater than 25 mmHg at rest. It is important to make an early diagnosis, because the prognosis is very poor (mean 5 year survival was 38.1%). The PH can primary result from the increased arteriolar resistance caused by luminal narrowing and/or constriction [1], elevated pulmonary venous pressure, and chronic pulmonary thromboembolism. The luminal narrowing is caused by the endothelial hyperplastic proliferation with the granular fibrin production, encroaching upon the lumen with thrombi organization. This reaction leads to the increased fibrinolytic and phagocytic activity of endothelial cell of pulmonary artery [2,3]. Consequently, concentric intimal fibrosis and so-called plexiform lesions which represent angio-proliferative aspects distinct from atherosclerosis are formed. PH also results secondarily from increased PA blood flow, such as congenital right-left shunt heart disease. The major features of PH on CT are central pulmonary artery dilatation (PA trunk, the diameter of which frequently exceeds that of the ascending aorta; dilatation of the right and left main pulmonary arteries; right ventricular and atrial enlargement with inversion of the interventricular septum and dilatation of the tricuspid valve annulus. However, the detailed feature of the peripheral pulmonary vessels has not been elucidated on MDCT including thin slice high-resolution CT.
We studied 40 (17-82 mean 45.6-years-old, Female/Male = 24/16) patients with clinically verified pulmonary hypertension (PH), reviewing the pulmonary angiography (DSA), symptoms, HRCT, multiple-plane reconstructed (MPR) views and 3D-VRCT including contrast enhancement study. This study includes both idiopathic PAH (i-PH) (n = 20)and secondary PH (n = 20).The causes were as follows; CTEPH (chronic thrombo-embolic PH) (n = 3), collagen vascular diseases (MCTD 3, SjS 2, RA 2), ASD (n = 2), VSD (n = 2), aortic coarctation and PDA (n = 1), liver cirrhosis (n = 1), mitral valve regurgitation (n = 2), single atrium and single ventricle (n = 1), and Rendu-Osler-Weber disease (n = 1). In all cases, the mean PA pressure measured with right sided cardiac catheterization and/or estimated systolic pressure through tricuspid valve by echocardiography was higher than 25 mmHg. The plain CT scan (slice thickness 5 mm) with gapless HRCT (slice thickness 1-2 mm) and/or contrast enhanced (CE)-CT (slice thickness 3-5 mm with 3D-CT volume rendering images) were performed. The CT scans acquired on the Definition (flash) scanner (Siemens Germany) consisted of non-contrast sequence of 120 kV, 200 to 260 mA, 1.0-5 mm collimation, pitch of 1.5:1, and a bone reconstruction algorithm. Images were reviewed with both mediastinal (width, 400 Hounsfield units (HU); level, 40 HU) and lung (width, 1600 HU; level, -550 HU) windows. The consecutive HRCT with no slice gap has been introduced from 2012/10. MPR (multi-planner reconstruction such as axial, coronal, and sagittal) and 3D images were also obtained. The institutional review board of this study and the informed consents of all patients regarding with the purpose and risk of the CE-CT examinations were obtained.
The prominence of the pulmonary vasculature and/or tortuosity (n = 16) and dilatation of the central pulmonary arteries (n = 39) were frequently observed. The MPA/Ao diameter ratio and CTR (cardiothoracic rate) were 0.82-2.60 (mean 1.39), and 0.4-0.81 (mean 0.58), respectively. The pulmonary artery trunk diameter in the study group was 3.55 +/- 0.66 cm, main pulmonary artery/ascending aorta ratio 1.2 +/- 0.29. The RA (n = 27) (mean 48.6 mm) and RV (n = 31) dilatation (mean 41.3 mm) were accompanied. RV myocardial and papillary muscle hypertrophy was revealed in 4 cases (10%). The mosaic attenuation with/without consolidation (n = 13 32.5%) was observed mainly in CTEPH and patients with hemoptysis. The interlobular septal thickening (n = 18, 45%), arterio-venous fistula (n = 3, 7.5%) (Figure 1) with micro-aneurysmal dilatation (n = 2, 5%) (Figure 2), and centrilobular (perivascular) ground glass opacities (n = 6, 15%) were clearly depicted on HRCT. Bronchial artery dilatation (> 2 mm) was observed in 5 (12.5%) cases. In one case, trans pleural collateral arterial circulations from brachiocephalic, internal mammary, intercostal, inferior phrenic and coronary arteries were observed and their drain to the peripheral PV branches were confirmed with PA angiography. The aneurysmal dilatation of the peripheral PA might reflect the angiomatoid or plexiform lesion. Although, the tortuosity and dilatation of the pulmonary artery and arteriole were prominent, abrupt narrowing and tapering of the peripheral pulmonary vessels were not conspicuous, especially in secondary PH (Figure 3). The mediastinal/hilar lymphadenopathy was observed in 9 (22.5%) cases. Pleural and pericardial effusion were observed in 12 (30%) and 8 (20%) cases, respectively. The wall calcification of PA (n = 2) and right atrium (n = 1) was presumed to occur by the dystrophies of thrombus and/or vasculitis (Table 1). The mosaic perfusion was frequently seen in s-PH (CTEPH) (p < 0.05).On the contrary, the centrilobular GGOs were exceedingly rare finding in s-PH group.
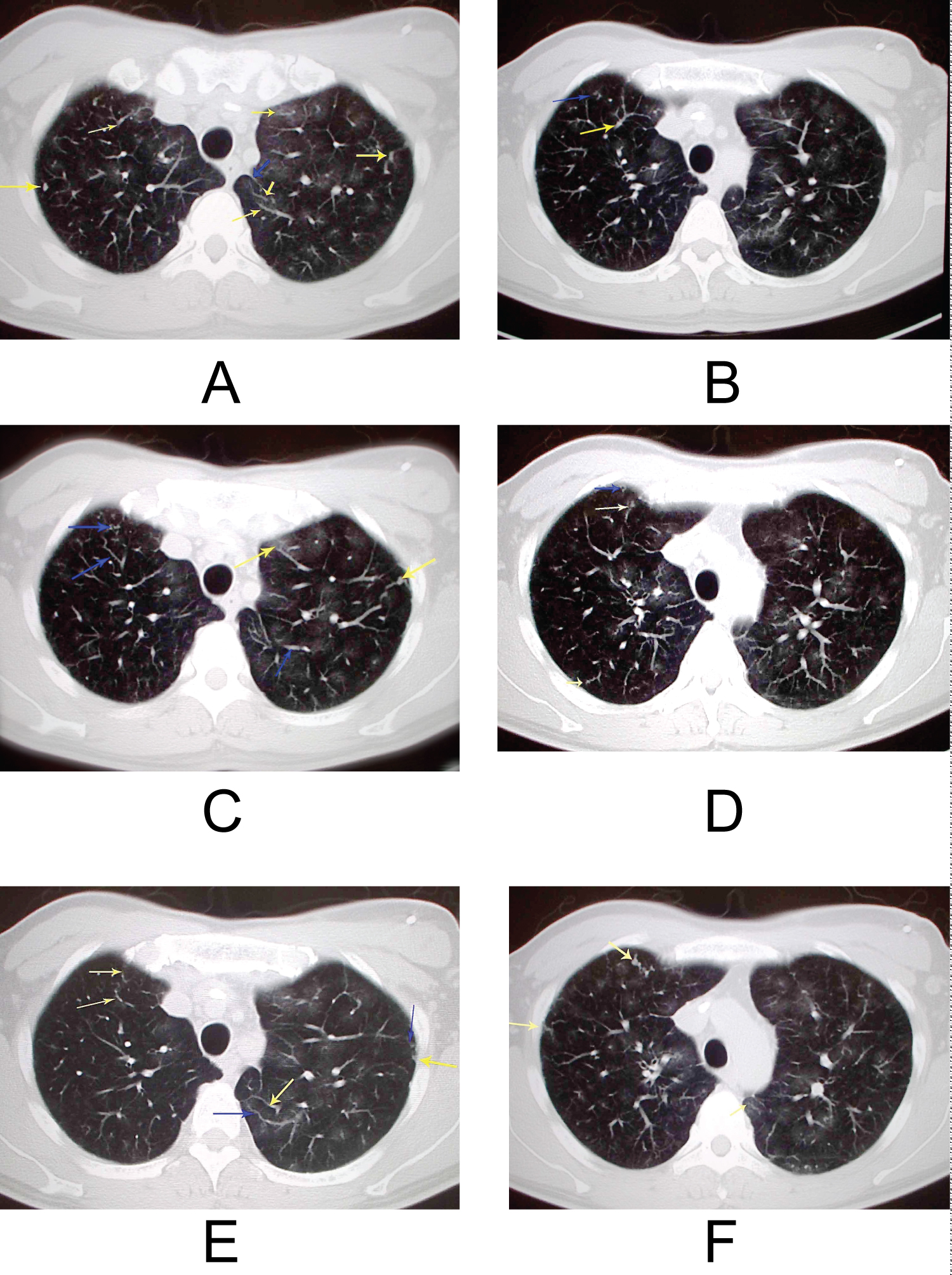 Figure 1: A 46-year-old male multiple AVF and centrilobular and perivascular ground glass opacities. PA pressure = 78/40/50 (mean/sys/diastolic) BP 129/75 PAR = 1.48 CTR = 0.55;
Figure 1: A 46-year-old male multiple AVF and centrilobular and perivascular ground glass opacities. PA pressure = 78/40/50 (mean/sys/diastolic) BP 129/75 PAR = 1.48 CTR = 0.55;
iPAH with multiple AVF and centri-lobular and perivascular ground glass opacities (a) Axial c HRCT scan obtained at the level of the apical segments of the upper lobes shows perivascular ground glass attenuation (arrows) in a predominantly centri-lobular distribution; (b-f) The multiple dilated peripheral vessels like AVF with cork-screw configurations were observed in both peripheral lung field.
Figure 1a-f: Yellow arrows: PA; Blue arrows: PV.
View Figure 1
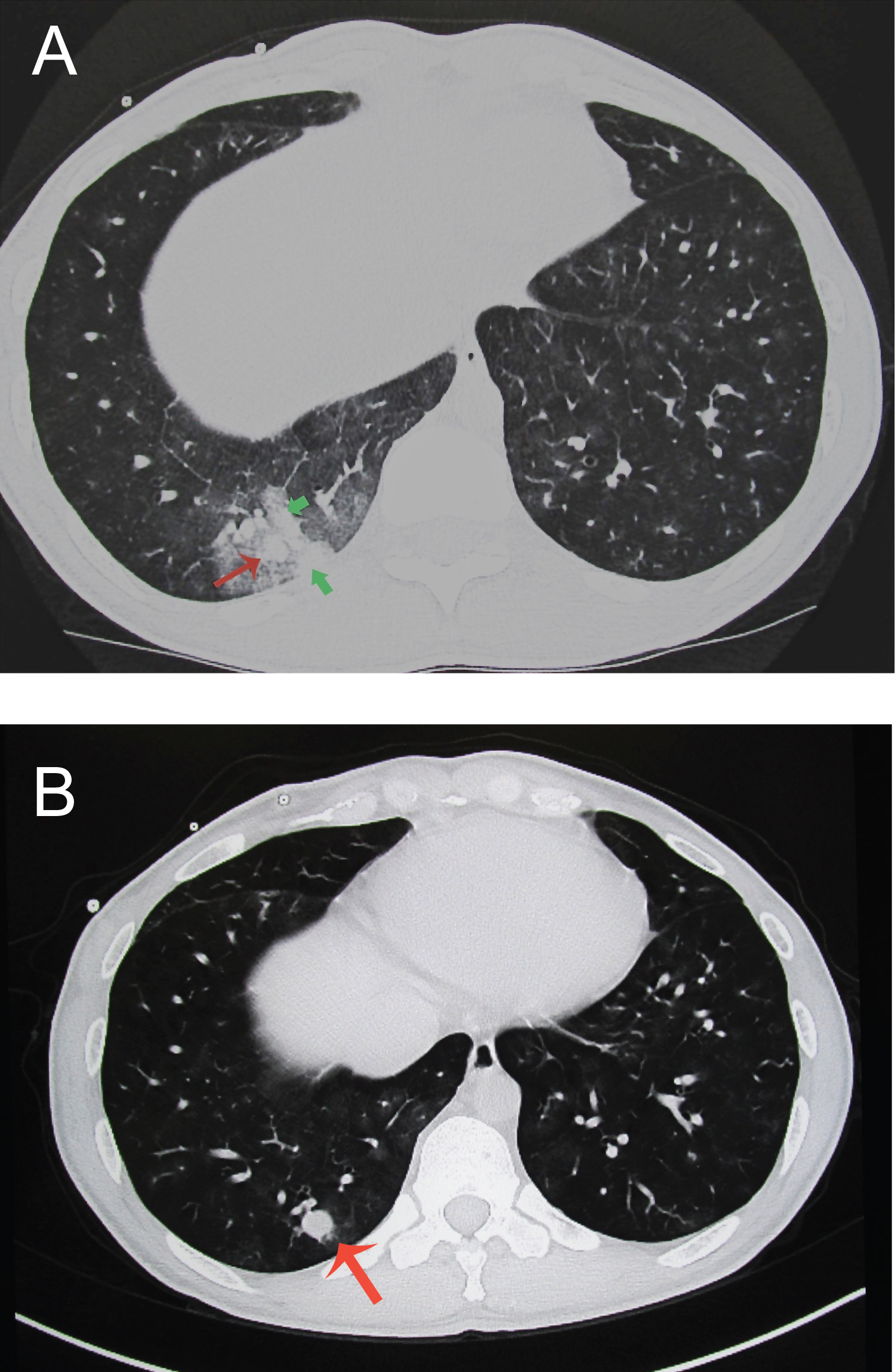 Figure 2: A 40-year-old man with i-PAH (mean PAP 60 (systolic 78/diastolic 40 mmHg BP104/52 (80) with several massive hemorrhage and the formation of aneurysmal dilatation in one month. He was treated with continuous infusion therapy of eboprostenol and the laboratory data showed microcytic anemia (Hb7.9 g/dl MCV 76.9 MCH 22.8) (a,b). The axial CT scan shows a pleura-based parenchymal high dense opacities (arrowhead) with ground glass opacity (a) in the right lower lobe (Segment 10). The size of A10 was 9.9 × 7.4 mm, after the pan-lobular opacities reflecting alveolar hemorrhage diminished, the growth of aneurysm was revealed (14 × 12.2 mm) (b). In both lung parenchyma, micro aneurysmal dilatation of peripheral PA with perivascular ground glass opacities were observed. After 3 months, the A10 aneurysm size grow up to the 20 × 14.5 mm (not shown) in diameter. The PAR and CTR were 1.41 and 0.40, respectively.
View Figure 2
Figure 2: A 40-year-old man with i-PAH (mean PAP 60 (systolic 78/diastolic 40 mmHg BP104/52 (80) with several massive hemorrhage and the formation of aneurysmal dilatation in one month. He was treated with continuous infusion therapy of eboprostenol and the laboratory data showed microcytic anemia (Hb7.9 g/dl MCV 76.9 MCH 22.8) (a,b). The axial CT scan shows a pleura-based parenchymal high dense opacities (arrowhead) with ground glass opacity (a) in the right lower lobe (Segment 10). The size of A10 was 9.9 × 7.4 mm, after the pan-lobular opacities reflecting alveolar hemorrhage diminished, the growth of aneurysm was revealed (14 × 12.2 mm) (b). In both lung parenchyma, micro aneurysmal dilatation of peripheral PA with perivascular ground glass opacities were observed. After 3 months, the A10 aneurysm size grow up to the 20 × 14.5 mm (not shown) in diameter. The PAR and CTR were 1.41 and 0.40, respectively.
View Figure 2
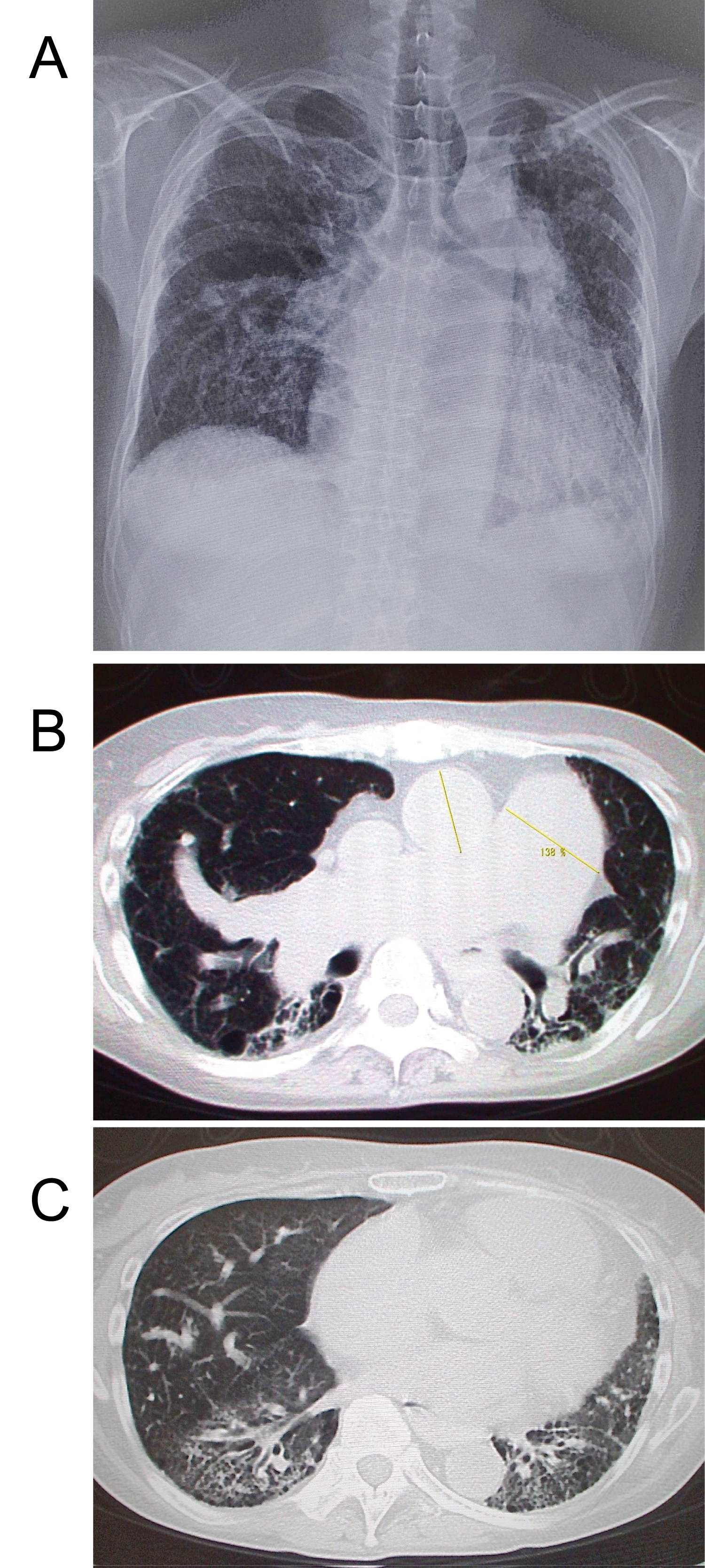 Figure 3: Secondary PH; A-52-year old female with MCTD PAR = 138%, CTR = 62% (a). The chest CXR showed prominent left 2nd arch and cardiomegaly with marked tortuosity of rt. middle lobe artery (b). MCTD-associated usual interstitial pneumonia were noted and estimated mean pulmonary artery pressure was 56 mm Hg on echocardiography. Axial CT scan obtained at the level of the carina shows dilatation of the pulmonary artery trunk and heterogeneous reticular opacities, subpleural honeycombing (c, arrows), traction bronchiectasis, and focal areas of ground-glass opacity.
View Figure 3
Figure 3: Secondary PH; A-52-year old female with MCTD PAR = 138%, CTR = 62% (a). The chest CXR showed prominent left 2nd arch and cardiomegaly with marked tortuosity of rt. middle lobe artery (b). MCTD-associated usual interstitial pneumonia were noted and estimated mean pulmonary artery pressure was 56 mm Hg on echocardiography. Axial CT scan obtained at the level of the carina shows dilatation of the pulmonary artery trunk and heterogeneous reticular opacities, subpleural honeycombing (c, arrows), traction bronchiectasis, and focal areas of ground-glass opacity.
View Figure 3
Table 1: The Summary of the CT (HRCT) findings in patients with PH (pulmonary hypertension). View Table 1
In PAH patients, Pulmonary artery and arterioles nearly always showed larger in size relative to the accompanied bronchi. Here, we discuss the new imaging findings of gapless HRCT and MPR reconstructed view including CE-CT. On CT, the central pulmonary artery dilatation, the diameter of which frequently exceeds that of the aorta; dilatation of the right and left main pulmonary arteries; right ventricular and atrial enlargement with inversion of the interventricular septum; corkscrew or tortuous configurations of peripheral pulmonary vessels indicative of plexogenic and dilative arteriopathy were clearly depicted. A mosaic pattern of attenuation and/or the centrilobular (vascular) ground glass opacities were also revealed. Plexogenic arteriopathy could occasionally be detected on CT, manifesting as irregular dilatation of PA. The centrilobular ground glass opacities clearly depicted on HRCT were assumed to reflect the perivascular hemorrhage or edematous reaction with neo-vascularity. The ILS (interlobular septum) thickenings were assumed to occur in the focal lymphatic or venous congestion area. The hilar/mediastinal lymphadenopathy (n = 9) were also assumed to reflect the lymphatic congestion or inflammation.
The size of atrium, right ventricle, and inferior vena cava (IVC) including the position of the interventricular septum can be evaluated more precisely on MDCT than echocardiography. The muscle hypertrophy were assumed to be caused by the chronic pressure overload [4,5] with/without volume overload subsequently.
The causes of precapillary (pulmonary arterial) PH include cardiac left-to-right shunt, chronic thromboembolic disease, and widespread pulmonary embolism arising from malignant cells, or parasites. Heath and Edwards classified the histopathologic features of PH in 1958. Attention focused on 6 grades of structural abnormalities in muscular pulmonary arteries and arterioles [6]. Moreover, malformed, tortuous vessels, and dilated, congested capillaries within alveolar septa, pulmonary arterial adventitia and within the walls of medium-size muscular PA [7] were also observed. Pulmonary venous hypertension may be caused by mediastinal fibrosis, pulmonary veno-occlusive disease, or a left atrial neoplasm, mitral valve stenosis/regurgitation, left ventricular failure, the combinations of the interlobular septal thickening, indistinct vascular margins, pleural effusion, and, occasionally, airspace opacities like Figure 1 have been reported to be characteristic CT features of pulmonary venous hypertension [2].
In most cases, peripheral pulmonary artery (PA) did not show pruning, the dilatation of PA with multiple arterio-venous fistulas (n = 3, 7.5%) were also seen. The suspected etiologies were as follows; a) The irregular dilatation of peripheral PA might represent the angiomatoid plexiform lesion (proliferative remodeling or congenital malformation); b) The thin-walled vascular channels of elastic and muscularis of peripheral PA plexus lesions and/or angitic or high flow hemodynamic effect could evolute aneurysmal dilatation; c) From a hemodynamic perspective, perturbation from an abnormally opening pulmonary valve or sheer stress resulting from shunt flow may induce remodeling and aneurysmal transformation of the vessel wall; d) The abnormal angiogenesis of the pulmonary vasculature development in 7 weeks gestation may also relate with the formation of micro anastomoses (AV fistula) (Figure 1 and Figure 2).
Pulmonary multiple AVF or AV malformation occur sporadically but are most commonly seen in association with Rendu-Osler-Weber-disease, an autosomal dominant disorder that is characterized by cutaneous and mucosal telangiectasia and arteriovenous malformations in other organs [8]. Diffuse pulmonary arteriovenous shunting is also seen in pregnancy, polysplenia syndrome, liver cirrhosis, and complex cardiac malformations [9], circulatory status with severe hypoxemia due to high shunt volumes, with the eventual development of PH. The characteristic vascular features manifests at CT as dilatation of the pulmonary arteries and veins with a weblike, reticular vascular pattern in the lung periphery and centrilobular vessel associated micronodules connected by arcade like, dilated vascular branches [10], whereas diffuse telangiectatic microshunt may be visible only at microscopic analysis. At histologic analysis, intrapulmonary vascular dilatation manifests as dilated precapillaries, direct arteriovenous shunts, and dilated pleural vessels.
Secondary PAH is created by the congenital heart disease and collagen vascular disease (CVD) PAH is most commonly secondary to the severe left-to-right shunts that remain undiagnosed and untreated for al long time, such as ASD, VSD, PDA and less frequently, transposition of the great vessels and truncus arteriosus. PAH associated with CVD is most commonly observed in systemic sclerosis, mixed connective tissue disease (Figure 3). This patient group is related to interstitial fibrosis, which is most commonly manifested histologically as nonspecific interstitial pneumonia [11], pulmonary vascular disease, or both. A study of PH in 722 patients with systemic sclerosis in the United Kingdom showed a prevalence of approximately 12%, and more than 10% of patients with severe PAH have an associated connective tissue disease, such as mixed connective tissue disease, polymyositis, dermatomyositis, and systemic lupus erythematosus [12]. PAH in this patient group is related to interstitial fibrosis, which is most commonly manifested histologically as nonspecific interstitial pneumonia, pulmonary vascular disease, or both. Vasculopathy may represent a complication of pulmonary fibrosis but also frequently occurs in the absence of significant interstitial lung fibrosis, and, characteristically, the severity of PH is not correlated with the degree of interstitial fibrosis depicted on high-resolution CT [13]. The comparison of patients with idiopathic and sPAH associated with connective tissue disease showed that later are mainly women, older, have a significantly lower cardiac output, and show a trend toward a shorter survival [14].
CTR in secondary PAH group was large compared with iPAH group statistically (Mann-Whitney U test p = 0.0088). The presence of s-PH in pulmonary disease (cor pulmonale) is an unfavorable prognostic sign. Patients typically present with signs and symptoms related to the specific underlying disease. Restrictive lung diseases associated with sPH include idiopathic pulmonary fibrosis, in which pulmonary hypertension is reported to occur with a prevalence as high as 46% [15]; secondary interstitial pneumonias due to connective tissue disease, sarcoidosis, vasculitis, drug toxicity also causes PAH.
The pathophysiologic mechanisms of pulmonary hypertension in obstructive and restrictive lung diseases include acute hypoxic vasoconstriction, vascular remodeling due to sustained alveolar hypoxia, loss of cross-sectional lung area caused by destruction of the alveolar capillary septa, compression of the alveolar vessels by increased intra alveolar pressure (COPD), and fibrotic compression or obliteration of the pulmonary vessels.
However, in patients with pulmonary fibrosis, pulmonary artery dilatation may occur in the absence of PH; the decrease of lung volume and increased lung recoil on vascular structures cause pulmonary artery dilatation. Devaraj, et al. [16] found that the ratio of the pulmonary artery diameter to the ascending aorta diameter is a more reliable marker of pulmonary hypertension in patients with fibrotic lung disease than the absolute main pulmonary artery diameter. When flow through the pulmonary vascular bed is increased, PAH can occur without a rise in vascular resistance in patients with ASD, AVSD, and PDA [17], an appearance produced by secondary small-vessel arteriopathy or by thromboembolic obstruction of the pulmonary arteries because of emboli originating in or passing through arteriovenous connections. Other frequent CT findings include small, rounded parenchymal opacities in the lung periphery with a dilated, tortuous feeding artery and an enlarged draining vein, features that represent CT-detectable macroscopic arteriovenous shunts [18]. Diffuse macroscopic arteriovenous shunting typically manifests at CT as dilatations of the pulmonary arteries and veins with a weblike, reticular vascular pattern in the lung periphery and centrilobular vessel associated micronodules connected by arcade like, dilated vascular branches [12,19]. Intrapulmonary vascular dilatation manifests as dilated precapillaries, direct arteriovenous shunts, and dilated pleural vessels.
The vasogenic mosaic attenuation (n = 13, 32.5%) were observed mainly in chronic thrombo-embolic PH, therefore, mosaic perfusion was frequently seen in s-PH (p < 0.05).
The dilatation of bronchial and systemic arteries were estimated to be caused by compensative blood flow by hypoxic pulmonary parenchyma due to the long standing PH [2]. In a study by Remy-Jardin, et al. [17], enlarged bronchial and non-bronchial systemic arteries were found in 73% of patients with CTEPH and in only 14% of patients with i-PAH. And non-bronchial systemic collaterals (notably, inferior phrenic, intercostal, and internal mammary arteries) are visible in as many as 45% of cases of CTEPH [18]. The Summary of the CT (HRCT) findings showed the significant difference of CTR between i-PAH and secondary PH group was observed on the Mann-Whitney U test (p = 0.0088). The mosaic perfusion was frequently seen in s-PH (CTEPH) (p < 0.05), and the centrilobular GGOs were exceedingly rare finding in s-PH group.
The limitations of our study include the small number of patients and the lack of a control cases. Moreover, this study included COPD, IP, and CPFE as background lung diseases, which have different impacts on cardiac morphology and function.
The prominence of the pulmonary vasculature and/or tortuosity and dilatation of the central pulmonary arteries were frequently observed in severe PH patients. RV myocardial and papillary muscle hypertrophy were observed in 4 (10%) cases. CTR in secondary PH group was larger than i-PH group statistically. The peripheral PA aneurysm and irregular dilatation of PA with PV fistula were observed in 2 (5%) and 3 cases (7.5%) on HRCT.