Alport syndrome is an inherited disease of collagen IV leading to progressive glomerular sclerosis associated with variable degrees of proteinuria and haematuria, sensorineural deafness and ocular abnormalities. Here we describe a novel mutation involving the COL4A5 gene leading to X-linked Alport syndrome (XLAS) in a Singhalese family of six children born to nonconsanguineous parents of Sri Lankan origin. The eldest 19-year-old male was found to have microscopic haematuria and proteinuria. His renal biopsy showed focal segmental glomerulosclerosis with no immune deposits. Electron microscopy revealed prominent irregular thinning and lamellation of the glomerular basement membrane, consistent with Alport syndrome. He also had bilateral sensorineural hearing loss. His mother was diagnosed to have end stage kidney disease while two of his sisters were also detected to have proteinuria and haematuria with normal kidney function. Whole exome sequencing of the proband showed a novel hemizygous variant (c.4119_4123dupGAGTA) in exon 47 of COL4A5 gene. The same variant was detected in his mother and two affected siblings. Thus, confirming the diagnosis of X-linked Alport syndrome in the four patients with renal involvement in this family.
Alport syndrome is an inherited disease of collagen IV leading to progressive glomerular sclerosis associated with variable degrees of proteinuria, haematuria, sensorineural deafness and ocular abnormalities [1]. Pathogenic mutations in collagen IV α3, α4, and α5 genes (i.e., COL4A3, COL4A4, and COL4A5) have long been recognized as causes of Alport syndrome [2]. These genes encode for key structural components of the glomerular basement membrane namely the type IV collagen peptide chains (α3, α4, and α5) [2,3]. Alport syndrome is attributed to three main inheritance patterns namely, X-linked, autosomal dominant and autosomal recessive. Mutations in COL4A3, COL4A4 are responsible for the autosomal dominant and autosomal recessive forms of the disease while COL4A5 gene variants are responsible for X-linked type [3]. Previously the senior author described a Sri Lankan family with autosomal recessive Alport Syndrome due to a novel mutation in the COL4A3 gene in the Sri Lankan population [4]. Here we describe a novel mutation involving the COL4A5 gene leading to X-linked Alport syndrome in the same population.
A 17-year-old female was incidentally detected as having proteinuria and haematuria with normal kidney function while being evaluated for a febrile illness. Her renal biopsy showed minimal changes on light microscopy, was negative for immune deposits and only showed subtle abnormalities in the glomerular basement membrane. Her mother (47 years) was a diagnosed patient with end stage kidney disease due to an unknown cause. A family screening was conducted which revealed urinary abnormalities in two other siblings. Her 19-year-old brother was found to have microscopic haematuria and proteinuria with a urine protein to creatinine ratio of 3.1 g/g creatinine and a serum creatinine of 1.3 mg/dl. His renal biopsy showed focal segmental glomerulosclerosis with no immune deposits. Electron microscopy revealed prominent irregular thinning and lamellation of the glomerular basement membrane, consistent with Alport syndrome (Figure 1a). The diagnosis was confirmed by Whole Exome Sequencing of his DNA, which revealed a novel hemizygous mutation (c.4119_4123dupGAGTA) in exon 47 of COL4A5 gene. Whole exome sequencing was performed as follows; Peripheral blood samples of the proband and the family members were collected to the EDTA tubes after obtaining written informed consent. Proband's genomic DNA was extracted from the blood leucocytes using QIAamp DNA Mini Kit, Qiagen according the manufacturer's protocol. Extracted DNA was subjected to whole exome sequencing on an Illumina HiSeq platform followed by library preparation using the Agilent Sure Select Human All Exon + UTR kit. The study was conducted at the Human Genetics Unit Faculty of Medicine, University of Colombo following a protocol approved by the Ethics Review Committee of the Faculty of Medicine Colombo.
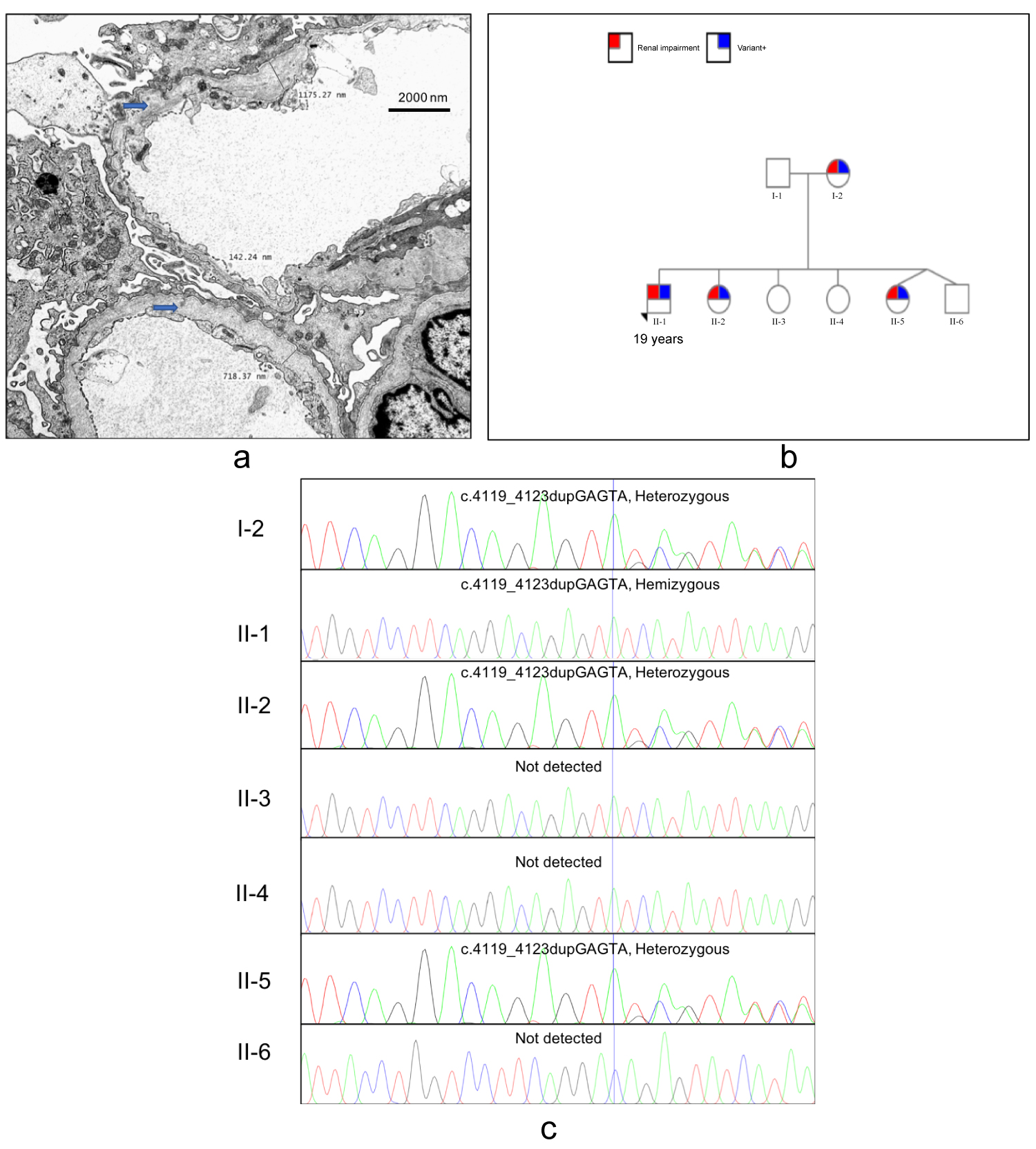 Figure 1: (a) Electron microscopy showing alternate thickening and thinning of the glomerular basement membrane with splitting or lamination of the lamina densa. (Uranyl acetate and lead citrate, original magnification x5000); (b) Pedigree showing the mutation and renal involvement; (c) Sanger sequence chromatogram results from family screening.
View Figure 1
Figure 1: (a) Electron microscopy showing alternate thickening and thinning of the glomerular basement membrane with splitting or lamination of the lamina densa. (Uranyl acetate and lead citrate, original magnification x5000); (b) Pedigree showing the mutation and renal involvement; (c) Sanger sequence chromatogram results from family screening.
View Figure 1
Genetic analysis of the paired end sequencing data was performed using an in-house bioinformatics pipeline. Obtained FASTQ files were mapped with the GrCh37 human reference sequence using BWA-mem algorithm and Genome Analysis Tool Kit (GATK) and the annotation of the VCF file was performed using SNP-eff with Refseq, clinical databases and population frequency databases. The variant calling was done using a virtual gene panel consisting of genes which were known to be associated with Alport Syndrome; COL4A3, COL4A4, COL4A5, COL4A6 and NPHS2. The identified variant was re-confirmed in the proband and the family members were screened for the identified variant by Sanger sequencing.
He was subsequently detected as having bilateral sensorineural hearing loss without any ocular abnormalities. Family screening revealed that his mother, 17-year-old sister and another 8-year-old sister having the same genetic mutation (Figure 1b and Figure 1c). The 8-year-old sister had proteinuria, microscopic haematuria with normal kidney function.
In this family, Alport syndrome is X-linked. Female patients with the X-linked disease are known to be less severely affected. However, the exact reason for this is uncertain. It is postulated that random X-inactivation [5] resulting in somatic mosaicism contribute to this phenotypic variability in females. In some studies it was observed following immunostaining for α5 Collagen IV in the glomerular basement membrane females affected with XLAS showed a variable degree of severity as compared to hemizygous male patients who showed a more severe renal manifestations while both groups carried the same genetic variants [6,7]. This difference in severity could be due to the presence of two X chromosomes in female carriers of XLAS [8]. With the advent of next- generation sequencing it is now possible to improve mutation screening in familial hematuric nephropathies [9,10].
Genotype to phenotype correlation of COL4A5 gene variants in females shows a 12% probability of developing end-stage renal disease or deafness by the age of 40 years. The risk increases after the age of 60 years with lifetime risk of progression to ESRD of approximately 25% [11]. Conversely in men XLAS shows a severer phenotype owing to the hemizygous nature of the X chromosome. The novel variant discovered appears to be of high pathogenicity since it has led to the development of ESRD in a female by the age of 47 years and both her daughters (17 years and 8 years) carrying the same variant had proteinuria and microscopic hematuria.
Collagen type IV α5 is composed of a single peptide chain a collagenous domain and a C terminal NC1 domain consisting of 14, 1414 and 224 amino acid residues respectively [9]. The variant identified c.4119_4123dupGAGTA is in exon 47 of COL4A5 gene. This gene has 51 exons and out of that 45 exons code for the collagenous domain [12]. Bioinformatics analysis of the variant predicted a substitution of Isoleucine amino acid, which is potentially pathogenic resulting in a frameshift mutation causing either a loss of the protein or a truncated protein due to a premature stop codon. This is a novel variant that has not been described before. Rare mutations in highly conserved areas are likely to be pathogenic. Due to the diversity in the inheritance pattern of COL4A3, COL4A4, and COL4A5 genes and their variable clinical expression, Whole Exome Sequencing (WES) is a powerful tool to understand the transmission pattern of Alport syndrome [13]. Thus, permitting clinicians to give timely genetic counseling keeping in mind the risk of recurrence. Genotype to phenotype correlation gives clinicians an in depth understanding of prognostic value of genetic testing in a large study conducted in 681 affected male participants with XLAS, 67% suffered from hearing loss and ocular changes were seen in 30%. It was also demonstrated that there exists a strong relationship between the position of the mutation and age at onset of ESRD [14].
Family history and clinical screenings of all family members still remains the gold standard for making a diagnosis of XLAS. Hearing loss and ocular changes have now been deemed more common than previously thought. In a study conducted in Germany it was found that a total of 39% from the study and more than 40% from literature of those who carried a variant had ocular changes [15,16]. Truncating mutations as seen in this family had two-fold greater odds of developing ocular complications compared to those with missense mutations. Hence these strong genotype-phenotype correlations help clinicians evaluate and counsel affected families with XLAS.
In conclusion this study confirms that female carriers of XLAS show a later onset of renal impairment and an overall less severe phenotype compared to males carrying the same genetic variants. Furthermore, the type of variant is also an indication of the prognostic severity of the condition pointing to the importance of variant identification in XLAS which can influence the clinical course of the disease.