Report of the first case of chondroblastoma involving the sinonasal cavity and ventral skull base, offering a description of the puzzling diagnostic process followed by its management. We describe the main radiologic and histologic characteristics of chondroblastoma and compare them to those of frequently mistaken differential diagnoses.
Case report of a 19-years-old male patient with final diagnosis of chondroblastoma.
A 19-years-old male patient presented with visual deterioration. Imaging revealed an expansile mass with an epicenter in the planum sphenoidale extending both into the sphenoid sinus and intracranially. The patient was managed with partial resection via endoscopic endonasal approach resulting in rapid recovery of vision.
Chondroblastoma of the skull base is an exceedingly rare tumor with varying prognosis. It is frequently confused with other bone tumors, such as chondrosarcoma and giant cell tumor. Meticulous radiological and histological review with high level of expertise is crucial in the diagnostic process. Complete resection is the mainstay treatment; however, lesions of the skull base should always be tempered with potential complications.
Chondroblastoma, Endoscopy, Skull base, Bone tumor
Chondroblastoma is a rare neoplasm of the bone constituting approximately 1% of all primary bone tumors. It is more common in men in their second or third decade of life and usually affects long bones. The skull base undergoes a mixed membranous and endochondral ossification; thus, it is rarely affected by chondroblastomas. Nonetheless, they most commonly affect the temporal bone [1,2]. This report presents the first case of a ventral skull base chondroblastoma.
For a period of two years, a 19-years-old healthy, African American male noticed progressive blurriness of his left eye with continual periods of aggravation associated with a pounding headache that would abate spontaneously after one or two hours. However, the patient did not seek medical attention until he suffered a motor vehicle accident associated with loss of consciousness. The patient reported a history of migraine headaches since childhood; however, he denied double vision, nosebleeds, nasal obstruction, or memory lapses and his family denied any evidence of personality changes (before or after the accident).
A brain and cervical spine computed tomography (CT) scans (Figure 1) showed a large expansile mass centered in the planum sphenoidale extending both intracranially and into the sphenoid sinus with involvement of the left anterior clinoid process. The outer margins of the mass demonstrated cortical bone that appeared predominantly intact except in a few areas of discontinuity. The mass measured 4.9 × 3.0 × 4.2 cm and had a soft tissue density center with Hounsfield Units varying from 8 to 115 (average of 55) as well as several areas of amorphous calcifications peripherally.
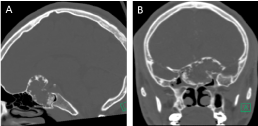 Figure 1: Preoperative CT scan A) Coronal; B) Sagittal (see text). View Figure 1
Figure 1: Preoperative CT scan A) Coronal; B) Sagittal (see text). View Figure 1
MRI showed that the central component of the lesion was isointense to hypointense on T1-weighted images relative to gray matter and heterogeneous on T2-weighted images. There were areas of extremely low T2 signal intermixed with cystic-appearing areas. Following intravenous administration of gadolinium, the lesion demonstrated heterogeneous but predominantly avid enhancement. The mass extended upwards into the intracranial fossa with mild displacement of the inferior portion of the left frontal lobe. No significant signal abnormality was noted in the adjacent brain parenchyma (Figure 2).
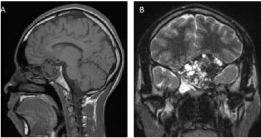 Figure 2: Preoperative MRI A) T1 sagittal MRI; B) T2 coronal MRI (see text). View Figure 2
Figure 2: Preoperative MRI A) T1 sagittal MRI; B) T2 coronal MRI (see text). View Figure 2
A preliminary diagnosis of an osseous lesion with a high suspicion for an ossifying fibroma was made, and the patient was subsequently referred to us for further management. This included a subtotal resection of the tumor with left optic nerve decompression via endoscopic endonasal approach (EEA). Intraoperative findings included a very large fibro-osseous lesion invading the sphenoid sinus, involving the sella and suprasellar space, and extending into the anterior skull base overlying the planum sphenoidale. In the context of a benign lesion, a subtotal extradural resection was done decompressing 270° of the left optic nerve canal and avoiding a CSF leak (Figure 3). The skull base defect was reconstructed using a right pedicled nasoseptal flap, which served to reline and protect the left optic nerve and internal carotid artery.
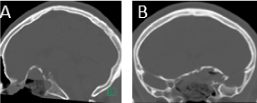 Figure 3: Post-operative CT scan showing resection of the center of the lesion with overall preservation of the peripheral osseous rim A) Sagittal; B) Coronal.
View Figure 3
Figure 3: Post-operative CT scan showing resection of the center of the lesion with overall preservation of the peripheral osseous rim A) Sagittal; B) Coronal.
View Figure 3
The patient was discharged with no complaints one day after surgery already noticing visual improvement. He followed an uneventful postoperative course and his left eye vision regained normal function within two weeks. The patient returned to his daily activity and continued doing well at 2 months of postoperative follow-up. After discussion of further treatment alternatives, the patient agreed to follow the residual tumor with imaging and monitor his vision with periodic neuro-ophthalmologic examinations.
The specimen consisted of multiple bony tissue fragments that measured 4.5 cm in greatest dimension. Light microscopic examination showed a solid proliferation of polygonal cells infiltrating among bone trabeculae. Proliferating cells had uniform rounded, ovoid and reniform hypochromic nuclei, evenly dispersed nuclear chromatin with occasional nuclear grooves but lacking nucleoli, a moderate amount of cytoplasm, and sharp cell borders. Mitotic figures were conspicuously infrequent, and necrosis was absent. Eosinophilic stromal matrix varied among tissue fragments with some being almost completely cellular (Figure 4A) while other fragments contained an excessive amount of hyalinized matrix (Figure 4B). Osteoclastic giant cells were scattered throughout the tissue. Immunostain results showed rare S-100 staining and diffuse positivity with vimentin.
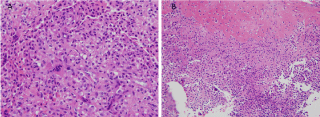 Figure 4: A) A solid sheet of mononuclear cells displays uniform cells with ovoid and reniform nuclei, discrete cells borders, occasional giant cells, and minimal matrix. H&E stain, x40; B) A large amount of extracellular stromal matrix is present in the upper half of this image causing a wide separation of cells. The lower half imitates the cells seen in figure 1A. H&E stain. x20.
View Figure 4
Figure 4: A) A solid sheet of mononuclear cells displays uniform cells with ovoid and reniform nuclei, discrete cells borders, occasional giant cells, and minimal matrix. H&E stain, x40; B) A large amount of extracellular stromal matrix is present in the upper half of this image causing a wide separation of cells. The lower half imitates the cells seen in figure 1A. H&E stain. x20.
View Figure 4
Chondroblastomas are rare benign neoplasms affecting any bone with endochondral ossification. Most commonly, it affects the long bones of men in their second or third decade of life. Rarely, it can affect the skull base most commonly arising in the temporal bone. To our knowledge this is the first report of a ventral skull base chondroblastoma with intracranial extension.
Chondroblastomas are commonly mistaken for other lesions, benign and malignant. Therefore, their differential diagnoses include benign lesions such as giant cell tumors, osteoblastomas, enchondromas, chondromyxoid fibromas and aneurysmal bone cysts, while malignant lesions include chondrosarcoma and osteosarcoma [2]. Chondroblastomas can be confused with chondrosarcoma when there is an excessive chondroid matrix with hypercellularity, while ossification of the matrix may mimic osteoblastoma. Sinonasal ossifying fibromas typically present as a lesion with a fibrous center and a thick peripheral osseous rim. Intermediate to low T1 signal along with mixed T2 signal composed of low signal intensity areas (calcification or ossification) and high signal intensity areas (fibrous tissue) are commonly described. Areas of ossification tend to increase in size with patient's age in ossifying fibromas [3]. Ossifying fibromas may be indistinguishable from sinonasal fibrous dysplasia, particularly when fibrous areas are more prominent, although the classic ground-glass appearance centrally would favor the latter. Another differentiating feature is that ossifying fibromas are typically monostotic whereas fibrous dysplasia can be monostotic or polyostotic. Sinonasal osteomas on the other hand tend to be more solid with central bone density therefore allowing differentiation with the previously described entities [4].
The characteristic radiographic image seen in chondroblastoma of the temporal bone includes an eccentric radiolucency and sharply demarcated sclerotic margin with areas of calcifications; however, these are only noticed in third of cases [1,2].
A classic histologic appearance of chondroblastoma includes the presence of highly cellular tissue composed of round or polygonal chondroblast-like cells with distinct outlines, multinucleated giant cells and chondroid, osteoid-like or more fibrous matrix with focal calcification. Tumors can show the characteristic pattern of chicken wire-type laciform calcifications [1]. There is a wide range of histological appearances, which frequently leads to confusion in the diagnostic process. However, osteoclast-like giant cells are commonly identified in chondroblastomas [1,2].
In the case herein presented, the chondroblastoma was initially interpreted as an ossifying fibroma compressing the left optic nerve. Thus, the goals of the surgery were to decompress the optic nerve, provide tissue for diagnosis and debulk the tumor while avoiding unnecessary complications such as CSF leak. The patient's vision improved significantly immediately after surgery and was normalized within 2 weeks postoperatively. Given that the final pathology was consistent with a chondroblastoma, and that the patient is doing very well postoperatively, it was decided to observe the residual component of the tumor using serial CT and MRI scans, and to monitor his vision with repeat neuro-ophthalmological examinations.
The standard management of chondroblastoma of long bones is intralesional curettage and bone grafting. However, Kutz, et al. [5] showed evidence of higher recurrence rate in patients with chondroblastoma of the temporal bone who underwent curettage in comparison to complete radical resection. They also emphasized on preservation of vital neurovascular structures. Recurrence rate of chondroblastoma of long bones varies between 5.7-38% [6-10]. Factors associated with recurrence include: Duration of symptoms for less than 6 months [1], incomplete resection [5] and secondary aneurysmal bone cyst formation [6,7]. Radiotherapy to chondroblastoma carries the risk of degeneration into chondrosarcoma and metastasis, hence it should be saved only for inoperable cases [5,11-13].
Chondroblastomas of the skull base are exceedingly rare lesions. Radiologically, they can be confused with other fibro-osseous lesions. An experienced pathologist with a high index of suspicion should always review histopathology of chondroblastoma since its presentation is commonly misleading and can be confused with other benign and malignant lesions. Most importantly, chondroblastoma must be differentiated from chondrosarcoma and giant cell tumor. Chondroblastomas seem to be best treated by complete resection; however, in the skull base, this needs to be tempered by potential complications (Table 1).
Table 1: Clinical and histopathological differences between chondroblastoma, giant cell tumor and clear cell chondrosarcoma [7,14-17]. View Table 1
i. All financial and material support for this research and work: Financial support was provided by the Anatomy Laboratory Toward Visuospatial Surgical Innovations in Otolaryngology and Neurosurgery (ALT-VISION) at the Wexner Medical Center at OSU. The laboratory is self-funded by the tuition of hands-on courses.
ii. Any financial interests the authors may have in companies or other entities that have an interest in the information in the Contribution (e.g., grants, advisory boards, employment, consultancies, contracts, honoraria, royalties, expert testimony, partnerships, or stock ownership in medically related fields): None.
iii. No financial disclosures.
None.