Prolyl oligopeptidase (POP) is a cytosolic serine protease with prominent expression in the brain. Inhibition of this enzyme leads to cognition-enhancing and neuroprotective effects in animal models with cognitive deficits. However, the biological function of POP remains unknown. Although in the past it was though that its catalytic activity was responsible for its physiological role, lately it has been hypothesized that POP is involved in the inositol pathway and that it interacts with several proteins, including α-tubulin, thereby implying that its function may be related to protein-protein interactions. In this review, we analyze the destabilization of microtubules in neurological diseases such as schizophrenia, Parkinson's, Alzheimer's and Huntington's disease. Given the interaction of POP with α-tubulin, we discuss the relevance of this protease in the modulation of synaptic processes. In this context, we also examine the potential of POP as a promising target for the treatment of cognitive impairment.
Microtubules, Neurological disease, Prolyl oligopeptidase, Protein-protein interaction, Synapsis
Prolyl oligopeptidase (POP, EC3.4.21.26), also known as prolyl endopeptidase, is a cytosolic serine protease (81 kDa) that hydrolyzes post-proline bonds of small peptides (less than 30 residues long). It was discovered in the human uterus in the early 1970s [1] and was first described as an oxytocin-hydrolyzing enzyme and further characterized as a peptidase that cleaves small substrates involved in learning and mnemonic processes, such as substance P, neurotensin, arginine-vasopressin and thyrotropin-releasing hormone, among others [2]. Some studies described that the administration of a POP inhibitor increases the levels of substance P and arginine-vasopressin in the hippocampus and frontal cortex [3]. However, it has been proposed that the hydrolytic activity of POP does not drive its physiological role, which remains elusive. In this regard, given that POP interacts with other proteins and it modulates the inositol triphosphate pathway, several alternative hypotheses have been put forward to explain POP function [4]. Despite further research is needed to elucidate the exact role of this enzyme in vivo, in this review, we focus on the known POP interactors and their relevance in POP function Regarding structure, POP is a monomeric protein that has an overall cylindrical shape formed by two domains. The catalytic domain is a typical α/β-hydrolase, whereas the structural domain is a seven-bladed β-propeller, which acts as an empty cylinder, restricting the size and orientation of the substrate [5,6] (Figure 1). Although the enzymatic mechanism of POP is still not fully understood, electron microscopy, NMR and X-ray crystallography studies have unveiled a state of equilibrium between open and closed conformations of this enzyme in solution. This equilibrium can be modulated by direct active site POP inhibitors [5,7].
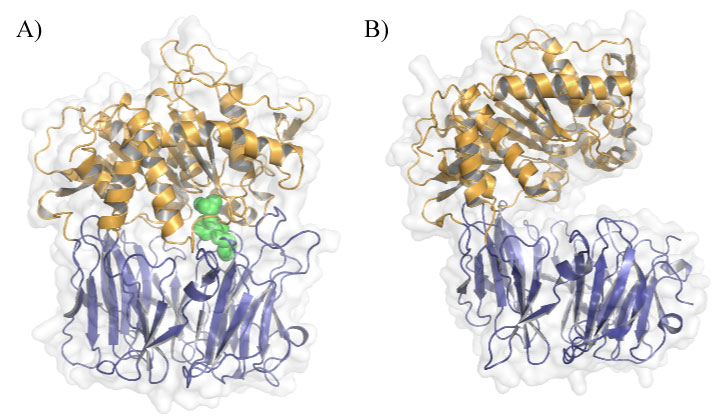 Figure 1: POP conformations in solution. A) Closed conformation of porcine POP (PDB ID: 1QFS) covalently bound to inhibitor Z-pro-prolinal (ZPP), the first POP inhibitor to be designed. β-propeller is shown in blue, α/β-hydrolase domain in orange and ZPP in green; B) Open conformation of Aemonas punctate POP (PDB ID: 3IUJ). View Figure 1
Figure 1: POP conformations in solution. A) Closed conformation of porcine POP (PDB ID: 1QFS) covalently bound to inhibitor Z-pro-prolinal (ZPP), the first POP inhibitor to be designed. β-propeller is shown in blue, α/β-hydrolase domain in orange and ZPP in green; B) Open conformation of Aemonas punctate POP (PDB ID: 3IUJ). View Figure 1
POP is ubiquitously expressed in the human body, but there is an increased concentration in the central nervous system (CNS) [8,9]. In this regard, this enzyme is expressed in cortical and hippocampal glutamatergic neurons, in ɣ-aminobutyric acid (GABA) ergic and cholinergic interneurons of the thalamus and cortex, and in Purkinje cells [10,11]. Moreover, after an inflammatory insult, POP expression is elevated in microglia and astrocytes [12].
Given the expression of POP in the CNS, its involvement in neurodegenerative and neuropsychiatric diseases has been addressed. Several studies report that POP activity is altered in patients with Alzheimer's disease (AD), Lewy body dementia, Parkinson's disease (PD), Huntington's disease (HD), and schizophrenia, among others [9]. POP activity has also been found to be altered in the serum of patients with mood disorders, such as depression and bipolar disorder [13]. However, care must be taken when interpreting these studies since many of them measured POP in plasma or post-mortem brain samples. In addition, experimental data show that POP inhibition has neuroprotective, anti-amnesic and cognition-enhancing properties in scopolamine-treated rats, whereas it decreases extracellular acetylcholine concentrations in the cortex and hippocampus of these animals [14,15]. The cognition-enhancing properties of POP inhibitors have been further demonstrated in other animal models with cognitive impairment [16-18] and in healthy human volunteers [19-22]. Furthermore, in cortical and cerebellar granule cells, POP inhibitors exert neuroprotective effects [23,24]. Three of these inhibitors, namely S-17092, JTP-4819 and Z-321, reached clinical stages for the treatment of AD [19-22]. However, unfortunately, the development of these drugs did not progress beyond these stages.
The presence of POP in the cytosol, together with its conformational dynamics and capacity to interact with other proteins, has led to the hypothesis that its biological function is related to protein-protein interactions. This notion was first put forward by Schulz, et al. in 2005, who proposed that POP regulates a number of cell functions independently of its peptidase activity [25]. In addition, it has been observed that POP inhibitors induce changes in the tertiary structure of the enzyme and can modify its interactome. The main POP interactors described in the literature are the cytosolic proteins α-synuclein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), growth-associated protein 43 (GAP43) and α-tubulin.
The abnormal fibrillary aggregation of α-synuclein protein in the cytoplasm of neurons and glia is the main characteristic of synucleinopathies, including PD, Lewy body dementia and multiple-system atrophy, among others [26]. The contribution of POP to enhancing α-synuclein aggregation was reported for the first time by Brandt, et al. in 2008 [27]. Since then, it has been shown that POP co-localizes with α-synuclein in the substantia nigra of PD patients [28] and later that the two proteins directly interact. This interaction was revealed through microscale thermophoresis and surface plasmon resonance studies [29]. In addition, POP inhibition reverses α-synuclein aggregation and promotes autophagy-the catabolic pathway used for clearing insoluble α-synuclein oligomers and fibrils [30]. These findings suggest that modulation of the α-synuclein-POP interaction using POP inhibitors could provide a new approach for the treatment of synucleinopathies.
GAPDH is a glycolytic enzyme involved in energy production pathways. However, this protein also participates in other non-glycolytic processes, including programmed cell death [30]. In this regard, it has been shown that GAPDH translocation and accumulation in the nucleus serves as an initiation signal for apoptosis [31,32]. Puttonnen, et al. and some years later Matsuda, et al. reported that POP inhibitors prevent GAPDH translocation into the nucleus under oxidative stress [33,34]. Later on, using co-localization, co-immunoprecipitation and proximity ligation experiments, Matsuda, et al. confirmed the interaction of POP with GAPDH [33]. This information strongly supports the notion that POP mediates GAPDH nuclear translocation and, consequently, that it may be involved in the regulation of apoptosis.
Among other CNS-related functions, GAP43 is involved in growth cone formation and axon guidance [35,36], although the underlying molecular mechanism is still not well understood. Valproic acid, carbamazepine and lithium treatment induce an increase in the average spread area of growth cones in sensory neuron cultures, and this increase is reversed by the administration of POP inhibitors [37]. This finding suggests that POP is also related to growth cone development. In this regard, Di Daniel, et al. proposed that this function of POP might be explained by its interaction with GAP43. They demonstrated binding between POP and GAP43 using co-immunoprecipitation and yeast two-hybrid assays [38]. Years later, this study was questioned by Szeltner, et al. who reported partial co-localization of POP and GAP43, without strong physical binding. In this regard, those authors reported a weak and transient interaction between the two proteins, as demonstrated by a glutaraldehyde cross-linking assay [39]. Despite this contradiction, it is plausible that POP participates in growth cone development via direct or indirect interaction with GAP43.
Repeated assemblies of α- and β-tubulin heterodimers comprise the structure of microtubules (MTs), which, together with microfilaments and intermediate filaments, form the cell cytoskeleton. Therefore, MTs are involved in a large number of cellular processes, including mitosis, cell motility, maintenance of cell shape, neurite growth, intracellular transport and vesicle secretion [40]. Using co-localization and yeast two-hybrid assays, Schulz, et al. demonstrated POP binding to the C-terminus region of α-tubulin [25]. They also showed that POP inhibition promotes protein and peptide release in U-343 cells. These results suggest that POP participates in MT-associated processes, such as intracellular trafficking, protein secretion and axonal transport.
Since several POP inhibitors have been demonstrated to improve memory and cognition in animals, the possible implication of POP in synaptic function has been studied using alternative approaches.
Firstly, in the same publication that proposed POP interaction with GAP43, the authors also generated POP knockout (KO) mice. These animals were used to demonstrate that the absence of POP causes a decrease in the number of collapsed growth cones and an increase in their spread area in explants when compared with wild-type (WT) mice [38]. Likewise, they observed that WT mouse explants treated with valproic acid, carbamazepine or lithium (all of them considered mood stabilizers) also show a decrease in the number of growth cones. However, treatment with POP inhibitors reversed the effect of these three mood stabilizers. Höfling, et al. also determined that POP KO mice show an increased expression of polysialylated-neural cell adhesion molecule (PSA-NCAM) [41], which has been associated with enhanced brain plasticity [42]. Alternatively, D'Agostino, et al. used POP gene trap mice to study the involvement of POP in synapsis. They observed that these mice present reduced hippocampal long-term potentiation, impaired learning and memory processes related to the hippocampus, decreased spine density in the hippocampus, especially in the CA1 region, and reduced GAP43 expression [43].
Given these observations, it is plausible that POP participates in the modulation of neuronal synapses. Although the molecular mechanisms underlying such modulation are not well understood, the interaction POP-GAP43 and POP-α-tubulin could provide an explanation. Here we outline some basic ideas about α-tubulin and, consequently, MT involvement in synaptic processes and in neurological diseases associated with POP dysregulation.
As mentioned previously, MTs are the major component of the cell cytoskeleton, and as such they are present in all eukaryotic cell types. The key properties of MTs are their dynamic behavior and directionality. The orientation of α- and β-tubulin heterodimers that make up the MT structure is particularly important for the polarity and stability of these cytoskeletal components. The (-) end of a MT has an exposed α-tubulin subunit, whereas the (+) end-where MT elongation takes place-has an exposed β-tubulin. Regarding neuronal MTs, which are characterized by the formation of the bundles required for the growth and maintenance of neurites [40,44], it has been described that axonal MTs are uniformly directed, with (+) ends orientated to axon tips, whereas in dendrites, MTs present mixed orientations (Figure 2). An additional difference between the MTs of axons and dendrites is their predominant microtubule-associated proteins (MAPs). Tau is found mostly in axons, while MAP2 predominates in dendrites. Despite these variations, there is general consensus that MT dynamics play a key role in ensuring the correct function of axons and dendrites, and consequently, in memory, learning and other cognitive processes. Indeed, MTs, in cooperation with other proteins, are involved in the regulation of intracellular cargo trafficking in axons and dendrites, as well as in the modulation of dendritic spine morphology and synaptic plasticity [45,46].
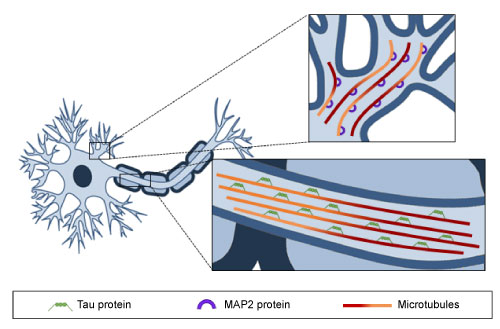 Figure 2: Microtubule distribution along axons and dendrites. MTs are oriented uniformly in axons and Tau protein is the predominant MAP, whereas in dendrites MTs show a mixed orientation and MAP2 protein is bound to them. View Figure 2
Figure 2: Microtubule distribution along axons and dendrites. MTs are oriented uniformly in axons and Tau protein is the predominant MAP, whereas in dendrites MTs show a mixed orientation and MAP2 protein is bound to them. View Figure 2
Neurodegenerative diseases are commonly characterized by a decrease in MT mass, as well as disrupted polarity patterns and impaired axonal transport, which are caused, in part, by a reduction in MT stability [40,47]. These findings support the notion that the modulation of MT dynamics could provide a potential therapy through which to improve the cognitive impairment associated with neurological diseases. Given the reported interaction between α-tubulin and POP and the implication of these proteins in synaptic processes, it cannot be ruled out that POP, and therefore POP inhibition, regulates MT dynamics (Figure 3). However, the interaction between POP and α-tubulin and the possible implication of this protease in MT stability requires further attention.
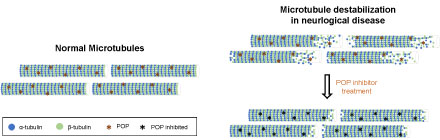 Figure 3: Destabilization of microtubules in neurological diseases. The diagram shows the difference in MT conformation between healthy subjects and patients with neurological diseases. The hypothetical recovery of the normal MT structure after POP inhibition is also shown. View Figure 3
Figure 3: Destabilization of microtubules in neurological diseases. The diagram shows the difference in MT conformation between healthy subjects and patients with neurological diseases. The hypothetical recovery of the normal MT structure after POP inhibition is also shown. View Figure 3
POP has been proposed to be a promising target for the cognitive impairment present in some CNS diseases, including schizophrenia, PD and AD, among others. This proposal has come about on the basis of three main observations: i) The brains of individuals with these conditions present altered levels of POP [13]; ii) The hypothesized implication of POP in spine density and formation [38,41-43] and; iii) The promising discoveries about molecules that interact with POP [25,29,33,38].
According to the WHO, schizophrenia is a neuropsychiatric disease that affects more than 1% of the population worldwide. Individuals with this condition can manifest three types of symptoms: Positive, negative and cognitive. Several antipsychotics for the treatment of positive and negative symptoms are available on the market. However, to date, no drug has been approved to treat cognitive impairment. POP emerged as a promising target to tackle the cognitive symptoms of schizophrenia [48]. Successful results regarding this indication were published by Prades, et al. In that study, three distinct mouse models of schizophrenia-which were based on the administration of subchronic phencyclidine (PCP) or acute dizocilpine (MK801), and on maternal immune activation induced by polyinosinic: Polycytidylic (PIC) during pregnancy-were treated with the POP inhibitor IPR19. Those authors showed that this inhibitor reversed the impaired cognitive symptomatology in a number of cognition tests that evaluate working and visual memory [49]. Therefore, it was concluded that POP inhibition exerts cognition-enhancing effects in animal models of schizophrenia.
Patients with schizophrenia show alterations in dendritic spines. These abnormalities have been observed in multiple brain regions, specifically in layer 3 of the neocortex, where pyramidal cells present a lower density of the smallest spines. It has been hypothesized that these spine deficits appear during early childhood and adolescence, probably as a result of disturbances in molecular mechanisms such as spine formation, pruning, and/or maintenance [50,51]. POP inhibitors may be able to restore dendritic spine deficits and therefore contribute to improving cognitive symptomatology.
PD is a neurodegenerative disorder characterized by loss of neurons in the substantia nigra and an accumulation of Lewy bodies, which comprise aggregated forms of α-synuclein. As previously mentioned, α-synuclein co-localizes and interacts with POP in the substantia nigra [28,29]. Nevertheless, no differences in POP activity or expression between PD and control subjects have been observed [28].
MTs are involved in PD, as demonstrated by observations of the interaction of tubulin with α-synuclein [52]. In addition, α-synuclein has been linked to MT stabilization, although the mechanism of action is not well understood. It has been proposed that α-synuclein is a MAP and that it therefore modulates the stabilization, polymerization and dynamics of MTs. Another hypothesis is that the interaction between α-synuclein and GSK-3β modulates Tau phosphorylation and consequently MT stabilization. Nevertheless, these two hypotheses are not necessarily exclusive [53].
Myöhänen, et al. demonstrated that POP is involved in α-synuclein aggregation. In this regard, those authors showed that POP inhibitor KYP-2047 stimulates the clearance of α-synuclein aggregates by enhancing macroautophagy in the A30P transgenic mouse model of PD [30].
The hallmark of AD is the accumulation of misfolded β-amyloid peptide plaques in the extracellular space, as well as the aggregation of phosphorylated Tau protein in neurons-a process that results in neurofibrillary tangles. The brains of AD patients show altered expression and activity of POP, and this protease has also been observed to co-localize with β-amyloid protein both intra- and extracellularly in this organ [9,29]. Tau protein co-localizes with POP inside cells in AD and control brains [26].
MTs are involved in the development of AD, as demonstrated by their reduced length and density [54], which indicate a possible alteration of synaptic processes. In this regard, in early stages of AD, β-amyloid alters changes in dendritic spine shape, whereas in later stages the cellular response to β-amyloid toxicity leads to a reduction in MT density and length and to the loss of dendritic spines [55].
HD is an inherited neurodegenerative disorder characterized by motor, psychiatric and cognitive deficits [56]. Cognitive impairment is one of the earliest symptoms of this disease. HD is caused by an abnormal CAG repeat expansion within exon 1 of the human huntingtin gene (HTT) [57], which encodes the huntingtin protein. Mutated huntingtin forms oligomers and globular intermediates that lead to aggregates, which in turn promote neuronal dysfunction and neurodegeneration [58].
HD is another example of a pathological process in which POP activity is decreased in brain samples of patients respect to controls [59]. Therefore, HD presents abnormalities in MTs and the cytoskeleton, most of these abnormalities being related to MAP dysregulation. Post-mortem HD brain samples present hyperphosphorylated Tau aggregates, a phenomenon observed in AD [60]. Dendritic atrophy induced by the dysregulation of MAP2, probably caused by splicing altered events, has also been reported [61].
Patients with neurological disorders, including AD, PD, HD and schizophrenia, present altered POP expression patterns. Moreover, the last two decades have brought about the design and testing of inhibitors of this serine protease for the treatment of the cognitive impairment related to these pathologies. POP inhibitors have widely demonstrated in vitro and in vivo cognition-enhancing and neuroprotective properties. However, although some of these drug candidates reached clinical stages, the biological mechanisms underlying the effects of POP are still unclear. Even though, the catalytic activity of the protease has been traditionally proposed as the function responsible for the biological activity of POP, a new promising hypothesis attribute the in vivo functionalities of this protease to its involvement in key physiologically relevant protein-protein interactions.
One of the most promising POP interactors is α-tubulin, a component of the heterodimer that forms the MT structure. In this regard, several neurological diseases are characterized by alterations in the stability and formation of MTs, as well as deficits in spine density.
On the basis of the findings reviewed herein, POP emerges as potential new target for the treatment of the cognitive impairment associated with some neurological diseases and psychiatric diseases. Research efforts should now be channeled into studying the binding of POP to α-tubulin, the potential involvement of POP in the cytoskeleton and neurite growth, and the effect of POP inhibitors as MT stabilizers.
N.T., A.A-Ll, R.P. and T.T. are employees of Iproteos, S.L. T.T. is founder of Iproteos, S.L.
N.T. and A.A-Ll. drafted the manuscript, R.P. and T.T. revised the manuscript. All the contributors revised, gave their approval to the final version of the manuscript, and agreed to be accountable for all aspects of the work.