

International Journal of Cancer and Clinical Research
Nitric Oxide Attenuates Hypoxia-Induced 5-FU Resistance of Oral Squamous Cell Carcinoma Cells
Eri Sasabe*, Ayumi Tomomura, Mayuko Hamada, Naoya Kitamura, Tomohiro Yamada and Tetsuya Yamamoto
Department of Oral and Maxillofacial Surgery, Kochi Medical School, Kochi University Kohasu, Japan
*Corresponding author:
Eri Sasabe, Department of Oral and Maxillofacial Surgery, Kochi Medical School, Kochi University Kohasu, Oko-cho, Nankoku, Kochi 783-8505, Japan, Tel: +81-88-880-2423, Fax: +81-88-880-2424, E-mail: yoshieri@kochi-u.ac.jp
Int J Cancer Clin Res, IJCCR-2-014, (Volume 2, Issue 2), Original article; ISSN: 2378-3419
Received: February 27, 2015 | Accepted: March 13, 2015 | Published: March 16, 2015
Citation: Sasabe E, Tomomura A, Hamada M, Kitamura N, Yamada T, et al. (2015) Nitric Oxide Attenuates Hypoxia-Induced 5-FU Resistance of Oral Squamous Cell Carcinoma Cells. Int J Cancer Clin Res 2:014. 10.23937/2378-3419/2/2/1014
Copyright: © 2015 Sasabe E, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Hypoxic environments in tumors induce expression of hypoxia-inducible factor (HIF). HIF contributes to the development of the malignant progression through the induction of various target genes. We previously reported that treatment with chemotherapeutic drugs and γ-rays enhances expression and nuclear translocation of HIF-1α under normoxic conditions; susceptibility of oral squamous cell carcinoma (OSCC) cells to the drugs and γ-rays is negatively correlated with expression of HIF-1α protein. In this study, we investigated the influence of a nitric oxide (NO) donor, DETA-NONOate, and nitroglycerin (glyceryl trinitrate, GTN) on susceptibility to 5-fluorouracil (5-FU) in OSCC cells under both normoxic and hypoxic conditions, as NO reportedly reduces HIF-1α accumulation. Treatment with various doses of DETA-NONO ate suppressed expression of both HIF-1α and HIF-2α remarkably under hypoxic conditions. Although susceptibility toward 5-FU was attenuated under hypoxic conditions, the combination with DETA-NONOate contradicted the suppressive effects of induced hypoxia by restraining HIF-1α and HIF-2α expression. Furthermore, growth rates of tumor xenografts implanted into nude mice were attenuated by treatment with a combination of 5-FU and GTN. These results suggest that the therapeutic application of NO donor to conventional chemotherapeutic agents may improve the response of OSCC.
Keywords
Hypoxia-inducible factor, Nitric oxide, Oral squamous cell carcinoma, 5-fluorouracil
Abbreviations
ARNT: Aryl Hydrocarbon Receptor Nuclear Translocator, CDDP: Cis-diamminedichloroplatinum, 5-FU: 5-Fluorouracil, GLUT-1: Glucose Transporter-1, GTN: Glycerol Trinitrate (nitroglycerin), HIF-1α: Hypoxia-Inducible Factor-1α, HIF-2α: Hypoxia-Inducible Factor-2α, NO: Nitric Oxide, OSCC: Oral Squamous Cell Carcinoma, PHD: Prolyl Hydroxylase, pVHL: von Hippel-Lindau protein, ROS: Reactive Oxygen Species, VEGF-A: Vascular Endothelial Growth Factor-A
Introduction
Hypoxic environments in tumors due to decreased vascular supply and increased energy demand of cancer cells contributes to development and progression of malignancies, and resistance to cancer therapy. The heterodimeric transcription factor Hypoxia inducible factor (HIF) is a master regulator that can adapt tumor cells to hypoxic conditions [1]. Overexpression of HIF has been observed in many different cancers, including head and neck squamous cell carcinomas, and is associated with increased patient mortality [2-4]. HIF consists of an oxygen-regulated α-subunit and a constitutively expressed β-subunit, also known as aryl hydrocarbon receptor nuclear translocator (ARNT). Hypoxic conditions within tumors promote stabilization of HIF-α by inhibition of the oxygen- and proline hydroxylase (PHD)-dependent enzymatic hydroxylation of proline residues and subsequent translocation to the nucleus. The two important HIF-α subunits, HIF-1α and HIF-2α, can transactivate overlapping numerous target genes involved in blood supply, energy production, growth/survival, and invasion/metastasis [5]. HIF also mediates resistance to anti-cancer drug through promotion of drug efflux, induction of autophagy, and inhibition of apoptosis, senescence, and DNA damage [6]. We have also previously shown that the endogenous level of HIF-1α in oral squamous cell carcinoma (OSCC) cells is inversely correlated with the susceptibility of OSCC cells to chemotherapeutic drugs and ionizing irradiation through the decrease of reactive oxygen species and promotion of drug efflux, and that the silencing of HIF-1α enhances the susceptibility of OSCC cells to chemotherapeutic agents [7]. HIF is thus recognized as an attractive target for the development of novel cancer therapies.
Recently, various anticancer agents that target HIF-1 inhibition, including heat shock protein 90 inhibitor, topoisomerase inhibitor, mTOR inhibitor, thioredoxin-1 inhibitor, histone deacetylase inhibitor, tyrosine kinase inhibitor and others have been reported [8-10]. Some of these inhibitors have been subjected to preclinical and clinical trials to estimate their usefulness in anti-cancer therapy. However, although these agents can lead to reduced HIF-1α mRNA or protein levels, HIF-1 DNA-binding activity or HIF-1-mediated transactivation of target genes, they do not act on HIF-1 specifically, and this lack of specificity increases the difficulty in attributing any anti-tumorigenic effects specifically to inhibition of HIF-1α. Furthermore, anti-tumor effects of HIF-1 inhibitors are sometimes very different between in vitro and in vivo and significant therapeutic effects are not expected.
Previous studies demonstrated that nitric oxide (NO) has anti-tumor effects and can reverse resistance toward various chemotherapeutic drugs in cancer cell lines induced by hypoxia [11-13]. NO reportedly acts as an inhibitor of HIF-1α by decreasing intratumor hypoxia and reducing HIF-1α accumulation. This was related to sustained PHD activity in the presence of NO, due to inhibition of intracellular oxygen consumption by inhibiting mitochondrial respiration, and subsequently increasing intracellular oxygen redistribution and oxygen availability in the cytoplasm [14-16]. Furthermore, NO donors such as nitroglycerin (glyceryl trinitrate, GTN) have been demonstrated to improve the effects of cancer therapy by inhibiting HIF-1α in solid cancers [17-19]. Therefore, we explored whether NO can show anti-tumor effects through inhibition of HIF signaling under both normoxic and hypoxic conditions in OSCC cells. We show here that NO donor contradicts the resistance toward 5-FU of OSCC cells induced by hypoxia through down-regulation of HIF-1α and HIF-2α expression.
Materials and methods
Drugs
DETA-NONOate was provided by Calbiochem (La Jolla, CA). Nitroglycerin (glyceryl trinitrate, GTN) was kindly supplied by Nippon-kayaku Co. (Tokyo, Japan). 5-fluorouracil (5-FU) was provided by Sigma-Aldrich, Inc. (Saint Louis, MO). Details of treatments are given in the legends of the figures.
Cell culture and treatments
OSCC cell lines (OSC-2, -4, -5 and -6 cells) were established in our laboratory from patients with OSCC and have been described previously [20]. OSCC cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Nissui Pharmaceutical Co. Ltd., Tokyo, Japan) supplemented with 10%(v/v) fetal bovine serum, 10mM glutamine, 100units/mL of penicillin, and 100μg/mL of streptomycin (Invitrogen Co., Carlsbad, CA). Under normoxic conditions (20% oxygen), OSCC cells were cultured at 37°C in a humidified 5% CO2/95% air atmosphere. Hypoxic conditions (1% oxygen) were produced in chambers filled with certified 1% O2, 5% CO2, and 94% N2 at 37°C.
Preparation of total cellular extracts and Western blot analysis
Cells were solubilized with ice-cold lysis buffer containing 1 %(v/v) Triton X-100, 50mM NaCl, 25mM Hepes (pH 7.4), 1mM EDTA, 1mM EGTA, 1mM phenylmethylsulfonyl fluoride (PMSF), 20μg/ml aprotinin, 10 μg/ml leupeptin and 1μg/ml pepstatin. To isolate the total cellular fraction, the lysates were centrifuged at 12,500×g for 15 min at 4°C and the clarified supernatants were used as total cellular extracts. Protein concentrations were determined using a BCA protein assay kit (Pierce Biotechnology Inc., Rockford, IL). Extracted proteins (50μg/lane) were separated by SDS-polyacrylamide gel electrophoresis, and transferred onto an Immobilon-P membrane (Immobilon, Millipore Corporation, Bedford, MA). Blocking was performed in Tris-buffered saline containing 5% (w/v) skim milk powder and 0.1 %(v/v) Tween-20. The membranes were probed with the following diluted antibodies (Abs): anti-HIF-1α monoclonal Ab (Transduction Laboratories, Lexington, KY) at 1:500, anti-HIF-2α polyclonal Ab (R&D Systems, Minneapolis, MN) at 1:200, and anti-β-actin monoclonal Ab (Santa Cruz Biotechnology, Inc.) at 1:1000. Detection was performed with an ECL system (Amersham, Piscataway, NJ).
RNA extraction and real-time quantitative RT-PCR analysis
Total cellular RNA was extracted using an RNeasy total RNA isolation system (Qiagen Inc., Valentia, CA), and was quantitated by measuring optical density at 260 nm. The extracted RNA (1μg) was added to 20μl reverse transcription buffer (50 mM Tris-HCl, pH 8.3, 50 mM KCl, 10mM MgCl2, 1 mMEDTA, 10μg/ml bovine serum albumin and 1mM DTT) with 10mM dNTP, 50 U RNase inhibitor, μg oligo dT primer, and 50U of avian myeloblastosis virus reverse transcriptase (all from Takara Biomedicals, Kyoto, Japan). This mixture was incubated at 42°C for 40 min and heated at 99°C for 5 min.
PCR used a master mix based on TaqMan universal PCR master mix and run on the ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA). Primers and the TaqMan probe for HIF-1α, HIF-2α, vascular endothelial growth factor-A (VEGF-A), glucose transporter-1 (GLUT-1), and β-actin were obtained from Applied Biosystems.
Cell proliferation assays and cell death assays
Cell proliferation was estimated by a BrdU incorporation assay kit (Roche, Mannheim, Germany). Cell death was quantitatively evaluated by double staining with Annexin V and propidium iodide (PI) (Sigma-Aldrich Inc.) according to the manufacturer's instructions. The cells were then analyzed on a FACScan cytometer using CELLQUEST software (Becton Dickinson, San Jose, CA).
Xenograft tumor model
Cells (2 × 105/0.05mL) were injected into the backs of 6-week-old male BALB/c nude mice subcutaneously (Japan Clea, Osaka, Japan); 28 days after the injection, mice were surgically implanted subcutaneously in the intrascapular area with mini-osmotic pumps (Alzet, Cupertino, CA) filled under sterile conditions in accordance with manufacturer's instructions, with either 200μl of GTN or isotonic saline. Pumps had a mean flow rate of 0.25μl/hr. Tumor size was measured with calipers and calculated volume as (length × width2) × 0.5. Mice were administered 10 mg/kg 5-FU intraperitoneally every three days and then sacrificed at day 21.
Immunohistochemical analysis
Tumor samples from mice were fixed in 10% buffered formalin and embedded in a paraffin block. Paraffin sections of 4μm thickness were obtained, deparaffinized, and dehydrated using a graded ethanol series. For immunostaining, sections were transferred to a 10-mmol/L citrate buffer solution (pH 6.0) and heated twice in a microwave oven for 10 minutes. Endogenous peroxidase activity was quenched by exposure of the sections to 0.3% H2O2 in methanol for 5 minutes. The sections were incubated overnight at room temperature with primary antibodies to HIF-1α (1:100, NB100-105; Novus Biologicals, Littleton, CO, USA), HIF-2α (1:100, NB100-122; Novus Biologicals), VEGF-A (1:100, JH121; Thermo Fisher Scientific, CA, USA), GLUT-1 (1:100, ab14683; Abcam, Cambridge, MA), and PECAM-1 (1:10000, M-20; Santa Cruz Biotechnology, Santa Cruz, CA). Thereafter, the sections were incubated with EnVision horseradish peroxidase-labeled polymer (Dako, Glostrup, Denmark) for 30 minutes, followed by development with diaminobenzidine + chromogen (Dako) for 5 minutes. Sections were then counterstained with hematoxylin staining solution and examined under a light microscope.
Statistical analysis
Results are expressed as the mean ± SEM. Differences were compared using Mann-Whitney's U-test. P < 0.05 was considered statistically significant.
Results
Influence of DETA-NONOate on HIF-1α and HIF-2α expression in OSCC cells under normoxic and hypoxic conditions
To examine the concentration-dependent effects of NO on expression of HIF-1α and HIF-2α, we treated four OSCC cells (OSC-2, -4, -5 and -6 cells) with DETA-NONOate, a slow (T1/2=20h) NO releaser; the relative expression of HIF-1α and HIF-2α was determined at increasing concentrations of the NO donor. OSC-2 and -4 cells expressed low levels of HIF-1α protein, and OSC-5 and -6 cells expressed high levels, under normoxic conditions. On the other hand, HIF-2α protein level was low or hardly detectable in all examined cells under normoxic conditions. Hypoxic treatments induced expression of both HIF-1α and HIF-2α remarkably in almost examined OSCC cells except for OSC-5 cells. Under normoxic conditions, treatments with DETA-NONOate have biphasic effects on the expression of HIF-1α. Protein levels of HIF-1α increased at low concentrations of DETA-NONOate (50–100μM) followed by a decrease at high concentrations (500μM). However, DETA-NONOate did not influence expression of HIF-2α. Under hypoxic conditions, all examined concentrations of DETA-NONOate decreased both HIF-1α and HIF-2α expression dose-dependently (Figure 1A). Expression of HIF target genes, including VEGF and GLUT-1, were induced under hypoxic conditions; treatment with DETA-NONOate decreased VEGF and GLUT-1 mRNA levels under both normoxic and hypoxic conditions. The suppressive effects were remarkable, especially under hypoxic conditions (Figure 1B). These results suggest that exogenous NO decreases HIF-1α, HIF-2α, and expression of their target genes, especially under hypoxic conditions.
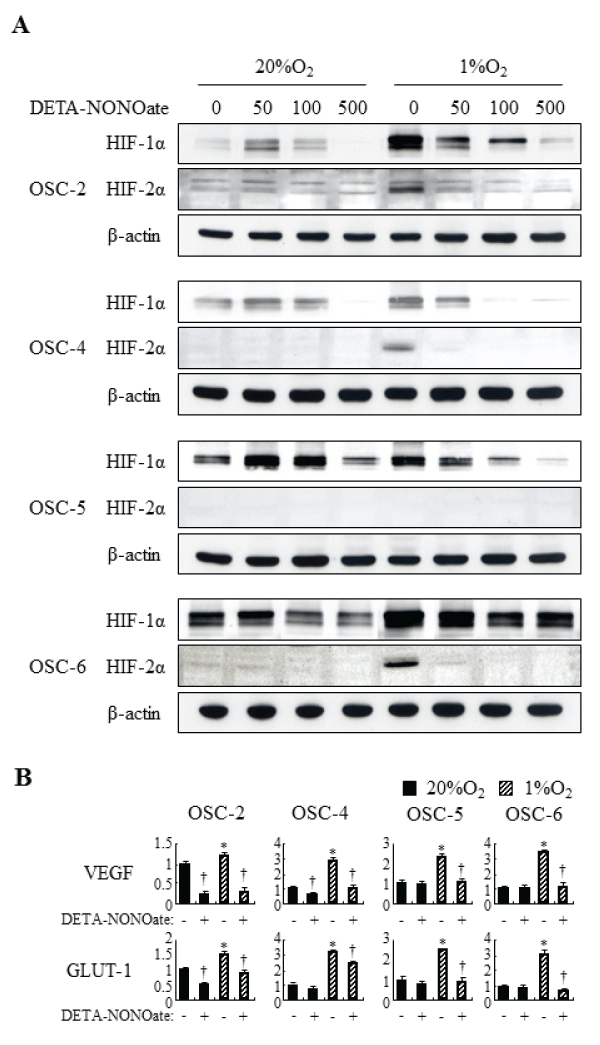
Figure 1: Effects of DETA-NONOate on HIF-1a, HIF-2a and expression of their target genes in OSCC cells under normoxic and hypoxic conditions.
(A) OSCC cells were incubated with increasing concentrations (50–500μM) of DETA-NONOate for 16 h. Total cell extract were prepared and processed by SDSPAGE
and western blot as described in Materials and Methods, using primary antibodies against: HIF-1a, HIF-2a, and ß-actin. (B) OSCC cells were incubated
with 500μM DETA-NONOate for 16 h. Expression of VEGF and GLUT-1 was assessed by quantitative RT-PCR. *: p< 0.05 against control cells under normoxic
conditions, †: p< 0.05 against cells without DETA-NONOate, by Mann-Whitney's U-test.
View Figure 1
Effects of the combination 5-FU and DETA-NONOate on HIF-1α and HIF-2α expression in OSCC cells under normoxic and hypoxic conditions
Because we previously reported that treatment with chemotherapeutic reagents including γ-rays, CDDP (cis-diamminedichloroplatinum), and 5-FU (5-fluorouracil) induced HIF-1α protein expression and nuclear translocation under normoxic conditions in OSCC cell lines, we next examined whether DETA-NONOate can contradict induction of HIF-1α by treatment with 5-FU in OSCC cells. Under normoxic conditions, expression of HIF-1α was induced by treatment with 5-FU (Figure 2). Conversely, the obvious inductive effects of 5-FU on expression of HIF-2α was not observed. On the other hands, treatments with 5-FU did not influence or rather decreased HIF-1α and HIF-2α protein levels slightly under hypoxic conditions. In either condition, expression of HIF-1α and HIF-2α were abolished completely by DETA-NONOate. These results suggest that NO inhibits HIF-1α expression induced by 5-FU under normoxic conditions, and DETA-NONOate can suppress HIF-1α and HIF-2α expression under hypoxic conditions potently.
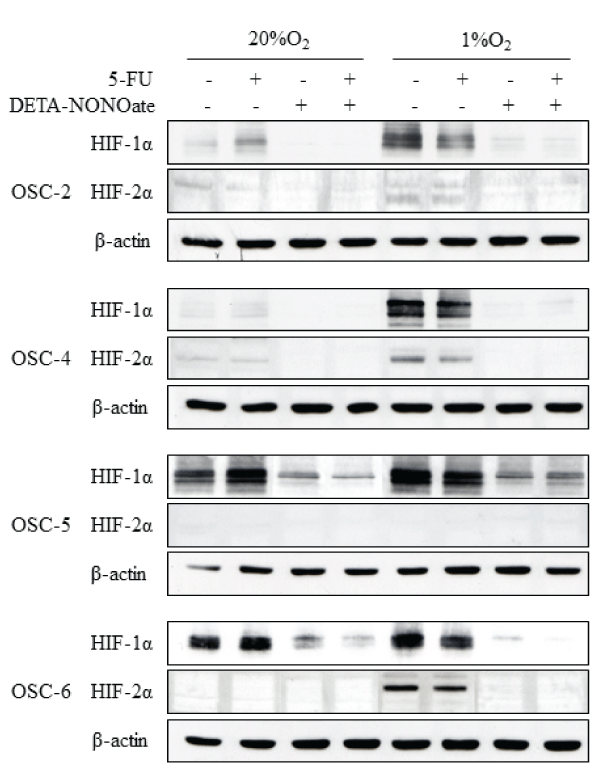
Figure 2: Effects of the combination of 5-FU and DETA-NONOate on HIF-1a and HIF-2a expression in OSCC cells under normoxic and hypoxic conditions.
OSCC cells were treated with 100μM 5-FU, 500μM DETA-NONOate, or their combinations and incubated under normoxic and hypoxic conditions. after 16 h,
total cell extract were prepared and processed by SDS-PAGE and western blot as described in Materials and Methods, using primary antibodies against: HIF-1a,
HIF-2a, and ß-actin.
View Figure 2
Effects of DETA-NONOate on susceptibility of OSCC cell lines to 5-FU
We first examined the effects of various doses of DETA-NONOate on cell proliferation. Low concentration DETA-NONOate (50μM) had no affect; intermediate concentration (100μM) inhibited it slightly. High concentration DETA-NONOate (500μM) suppressed proliferation of all cells about 40–50 % intensively under both normoxic and hypoxic conditions (Figure 3A). Although OSCC cells acquired resistance toward various doses of 5-FU under hypoxic conditions, cells treated with combined 5-FU and DETA-NONOate contradicted these effects. Furthermore, compared with normoxic conditions, the combination induced more obvious inhibitory effects on proliferation of OSC-2 and OSC-4 cells under hypoxic conditions (Figure 3B).
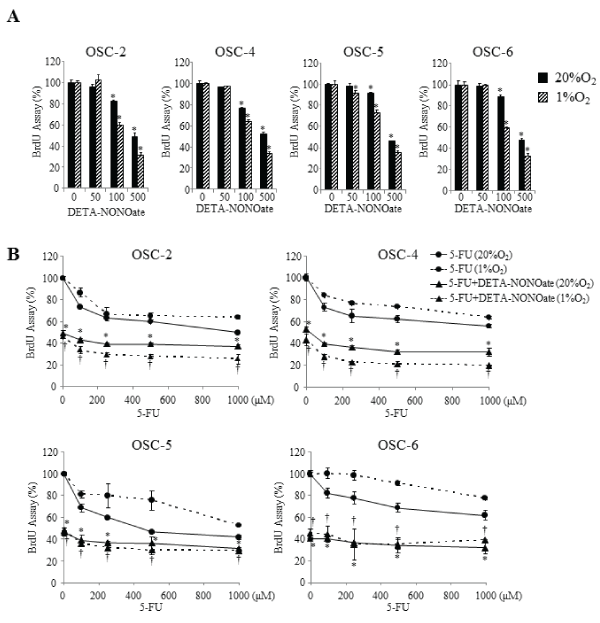
Figure 3: Effects of the combination of 5-FU and DETA-NONOate on cell proliferation.
(A) OSCC cells were incubated with increasing concentrations (50–500μM) of DETA-NONOate under normoxic and hypoxic conditions for 16 h. *P < 0.05
against control cells by Mann-Whitney's U-test. (B) OSCC cells were incubated with increasing concentrations (100–1000μM) of 5-FU in the presence of 500μM
of DETA-NONOate under normoxic and hypoxic conditions for 16 h. Cell proliferation was estimated by a BrdU incorporation assay. Data represent mean ±
SEM from three separate experiments. *: p< 0.05 against 5-FU treated cells under normoxic conditions, †: p< 0.05 against 5-FU treated cells under hypoxic
conditions, by Mann-Whitney's U-test.
View Figure 3
Effects of combination of 5-FU and DETA-NONOate on cell death in OSCC cell lines
We next examined the effects of DETA-NONOate on cell death in OSCC cells. Under normoxic conditions, low concentration DETA-NONOate (50–100μM) induced cell death slightly or had no influence, but high concentration DETA-NONOate (500μM) induced cell death strongly (Figure 4A). Conversely, under hypoxic conditions, the remarkable inductive effects on cell death were observed by treatments with all examined concentrations of DETA-NONOate. Although the number of dead OSCC cells decreased in various doses of 5-FU under hypoxic conditions, cells treated with combined 5-FU and DETA-NONOate contradicted these effects. Furthermore, as compared with normoxic conditions, the combination induced more obvious effects of cell death in all examined OSCC cells under hypoxic conditions (Figure 4B).
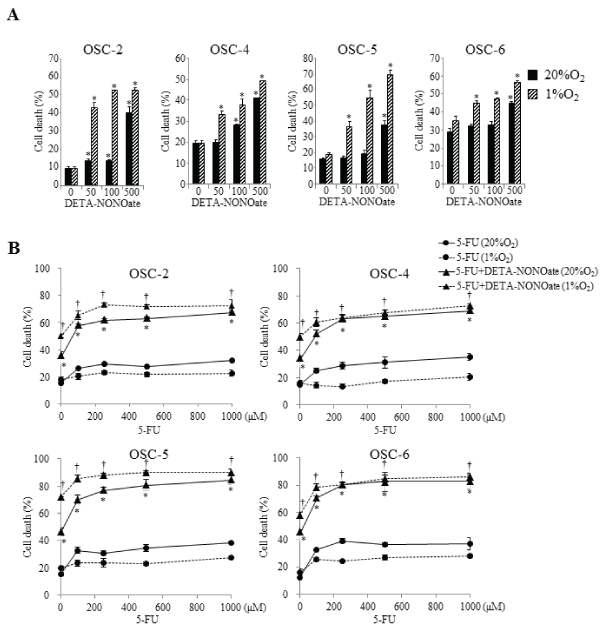
Figure 4: Effects of the combination of 5-FU and DETA-NONOate on cell death.
OSCC cells were incubated with 100μM 5-FU, increasing concentrations (50–500μM) of DETA-NONOate, or 100μM 5-FU plus 500μM DETA-NONOate under
normoxic and hypoxic conditions for 16 h. Cell death was quantitatively evaluated by staining with Annexin V and PI. The cells were then analyzed on a FACScan
cytometer using CELLQUEST software (Becton Dickinson, San Jose, CA). Data represent mean ± SEM from three separate experiments. *: p< 0.05 against 5-FU
treated cells under normoxic conditions, †: p< 0.05 against 5-FU treated cells under hypoxic conditions, by Mann-Whitney's U-test.
View Figure 4
Effects of GTN on tumor growth in xenograft models
To verify the antitumor effect of GTN in vivo, we constructed an endogenous animal model of OSCC cells by injecting OSC-4 cells into the back skin of nude mice. The treatment with GTN and 5-FU independently decreased the volume of both tumors. The combination of GTN and 5-FU showed the most prominent effects (Figure 5). Immunohistochemical analyses showed positive staining of HIF-1α, HIF-2α, VEGF and GLUT1 in control tumor cells. Their stains in the GTN-, 5-FU-, or combination-treated tumor cells were almost negative, but the peripheral tumor necrotic regions showed positive staining (Figure 6A). We also examined mRNA levels in the dissected tumors. Treatment with 5-FU and GTN independently decreased the mRNA levels of HIF-1α, HIF-2α and their target genes including VEGF and GLUT-1. However, the additive or synergistic effects were not shown in Figure 6B. Mean blood vessel density was also decreased by these treatments (Figure 6C).
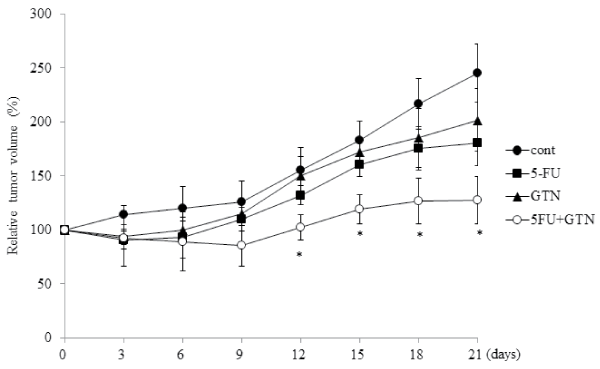
Figure 5: Effects of 5-FU, GTN, or its combination on tumor growth in xenograft models.
BALB/c nude mice were treated as described in Materials and Methods. Tumor volume of OSC-4 cells xenografted to back of mice was estimated every three
days for 21 days. *P< 0.05 against control tumor by Mann-Whitney's U-test.
View Figure 5
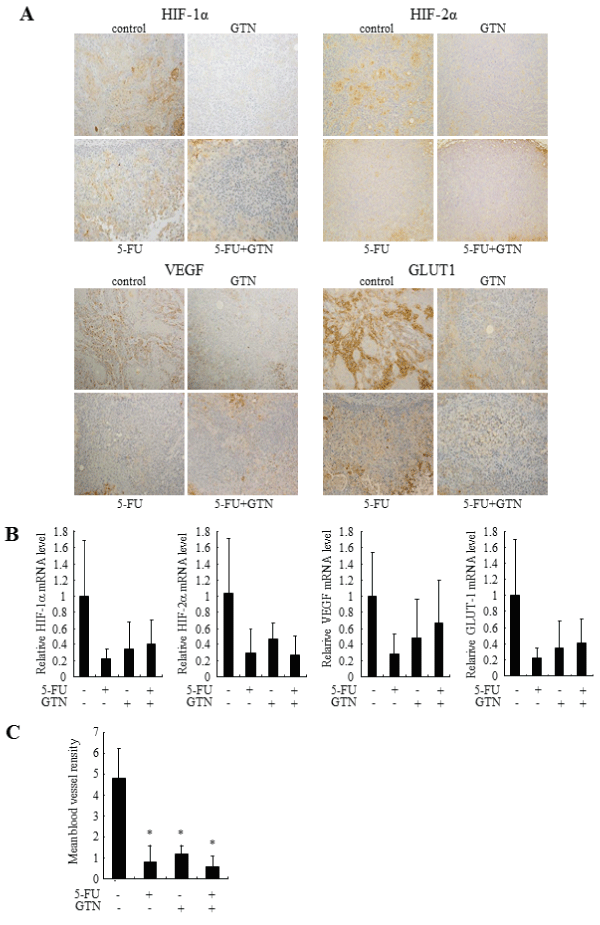
Figure 6: Effects of 5-FU, GTN, or its combination on HIF-1a, HIF-2a and expression of their target genes in xenograft models.
(A) Immunohistochemical appearance of OSC-4 tumor xenografted to BALB/c nude mice. Serial sections were stained with antibodies as described in Materials
and Methods. (B) On day 21, each tumor was collected and total RNA was purified. Expression of HIF-1a, HIF-2a, VEGF and GLUT-1 was assessed by quantitative
RT-PCR. (C) Serial sections were stained with anti-PECAM-1 antibody as described in Materials and Methods. Mean blood vessel density was determined by
estimating number of blood vessels per area. Data represent mean ± SEM from eight separate tumors. *P< 0.05 against control tumor by Mann-Whitney's U-test.
View Figure 6
Discussion
Hypoxia-induced HIF-1α and HIF-2α expression has been implicated in the induction of drug resistance to cancer cells [6]. NO is a ubiquitous molecule with diverse biological effects that depend on the source, concentration, latency, cell type and phenotype. A number of recent studies have provided evidence that NO plays an important role as a novel therapeutic to overcome tumor cell resistance toward chemotherapy and irradiation by activation of p53, induction of cytostasis and cell-cycle arrest through cyclin D1, and inhibition of drug efflux through multidrug resistance associated protein 3 [21-24]. Some NO donors, including nitroglycerin, sodium nitroprusside, and isosorbide dinitrate, were reported to inhibit HIF-1α accumulation and activation in hypoxic malignant cells, and to suppress growth of tumor cells [17,18]. In the present study, OSCC cells acquired resistance toward 5-FU under hypoxic conditions, but treatment with NO donors inhibited expression of HIF-1α and HIF-2α, especially under hypoxic conditions. Growth by OSCC cells was suppressed partially by inhibition of proliferation and induction of cell death. These results suggest that NO donors can down-regulate HIF expression and overcome chemoresistance induced by hypoxic environments in the tumor.
Several studies have implied that NO shows opposite effects on HIF-1α expression under normoxia or hypoxia; i.e., NO increased HIF-1α protein levels and HIF activity under normoxia and decreased them under hypoxia. Under normoxia, NO can inhibit PHD and factor inhibiting HIF (FIH) activity by interacting with enzyme-bound Fe2+, preventing hydroxylation of HIF-1α proline and asparagine residues that target HIF-1α for degradation and block interaction with the transcriptional co-activator p300-CBP [25-27]. Kasuno et al. reported that NO induced HIF-1 activation depending on MAPK and phosphatidylinositol 3-kinase signaling under normoxic conditions [28]. Conversely, during hypoxic conditions, competitive binding of NO to mitochondrial cytochrome c oxidase results in re-distribution of O2 to restore PHD activity [28]. NO can also inhibit HIF-1 activity by promoting cellular free iron and restoring PHD activity [29-31]. In the present study, under normoxia, low doses (50 and 100μM) DETA-NONOate stabilized HIF-1α and high doses (500μM) of DETA-NONOate suppressed HIF expression. However, all examined concentrations of DETA-NONOate inhibited HIF-1α and HIF-2α expression under hypoxic conditions. These results imply that regulation of HIF by NO depends on local O2 availability, NO concentration, NO metabolites or bioactive forms; and that hypoxia-induced HIF expression in tumor cells is always suppressed by treatment with NO donor.
Under normoxic conditions, we previously reported that chemotherapeutic drugs enhanced expression of HIF-1α, and these effects were abolished with treatment with the antioxidant N-acetyl-L-cysteine [7]. Chemotherapeutic drugs generate ROS in cancer cells, which impair proteins, lipids, and nucleic acids via strong oxidative activities [32-35]. Furthermore, we demonstrated that intracellular ROS produced by knockdown of Mn-SOD enhance HIF-1α expression by inhibiting degradation, and enhancing the transcription and translation, of HIF-1α [36]. These results suggested that chemotherapeutic drugs induced HIF-1α expression partially through the production of ROS under normoxia. In the present study, treating OSCC cells with 5-FU induced HIF-1α expression profoundly, whereas the expression was abolished in the presence of DETA-NONOate under normoxic conditions. Peroxynitrite (ONOO-) formed during interactions between NO and mitochondria derived ROS led the release of iron and 2-oxoglutarate (2-OG) from cellular compartment and caused a reduction of HIF-1α by increasing PHD activity [31]. The interaction of NO and ROS with PHDs may be relevant for the suppression of HIF expression. However, under hypoxic conditions, treatments with 5-FU decreased HIF-1α and HIF-2α expression in vitro and in vivo. Combination treatment with 5-FU and NO donor showed additive suppressive effects on HIF-1α and HIF-2α expression induced by hypoxia. These results suggest that the addition of NO donor to conventional OSC chemotherapies can improve the therapeutic effects regardless of normoxia or hypoxia.
HIF-1α and HIF-2α activate the expression of genes involved in proliferation, metabolism, angiogenesis, and metastasis. While highly homologous, HIF-1α and HIF-2α have been shown to have different roles in tumorigenesis, depending on specific tumor microenvironment. HIF-2α contributes to aggressive tumor phenotypes by activating hypoxia-inducible genes involved in tumor cell proliferation, angiogenesis, metastasis, and differentiation, promotion of EGFR translation, and crosstalk with c-myc [8,37]. In the present studies, HIF-2α expression was almost undetectable in OSCC cells under normoxia, but hypoxia increased its protein levels dramatically. Knockdown of HIF-2α by siRNA transfection inhibited cell proliferation under both normoxia and hypoxia (data not shown). Inhibitory effects on HIF-2α expression by an NO donor may contribute to tumor growth in addition to HIF-1α suppression.
Conclusions
In conclusion, the present study demonstrated that NO donor have antitumor effects even under hypoxic conditions and overcome hypoxia-induced chemoresistance in OSCC cells by modifying expression of HIF-1α and HIF-2α. It is important to develop novel therapeutic approaches using NO donors that target hypoxic tumor environments expressing abundant HIF-1α and HIF-2α. Therefore, it is necessary to further explore the details of NO-decreased HIF-1α and HIF-2α signaling in cancer cells.
Acknowledgements
This work was supported in part by Grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (20390480).
References
-
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721-732.
-
Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, et al. (1999) Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 59: 5830-5835.
-
Beasley NJ, Leek R, Alam M, Turley H, Cox GJ, et al. (2002) Hypoxia-inducible factors HIF-1alpha and HIF-2alpha in head and neck cancer: relationship to tumor biology and treatment outcome in surgically resected patients. Cancer Res 62: 2493-2497.
-
Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos C, Turley H, et al. (2002) Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int J Radiat Oncol Biol Phys 53: 1192-1202.
-
Gordan JD, Simon MC (2007) Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev 17: 71-77.
-
Rohwer N, Cramer T (2011) Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat 14: 191-201.
-
Sasabe E, Zhou X, Li D, Oku N, Yamamoto T, et al. (2007) The involvement of hypoxia-inducible factor-1alpha in the susceptibility to gamma-rays and chemotherapeutic drugs of oral squamous cell carcinoma cells. Int J Cancer 120: 268-277.
-
Lu X, Kang Y (2010) Hypoxia and hypoxia-inducible factors: master regulators of metastasis. Clin Cancer Res 16: 5928-5935.
-
Onnis B, Rapisarda A, Melillo G (2009) Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med 13: 2780-2786.
-
Xia Y, Choi HK, Lee K (2012) Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem 49: 24-40.
-
Matthews NE, Adams MA, Maxwell LR, Gofton TE, Graham CH (2001) Nitric oxide-mediated regulation of chemosensitivity in cancer cells. J Natl Cancer Inst 93: 1879-1885.
-
Frederiksen LJ, Siemens DR, Heaton JP, Maxwell LR, Adams MA, et al. (2003) Hypoxia induced resistance to doxorubicin in prostate cancer cells is inhibited by low concentrations of glyceryl trinitrate. J Urol 170: 1003-1007.
-
Frederiksen LJ, Sullivan R, Maxwell LR, Macdonald-Goodfellow SK, Adams MA, et al. (2007) Chemosensitization of cancer in vitro and in vivo by nitric oxide signaling. Clin Cancer Res 13: 2199-2206.
-
Huang LE, Willmore WG, Gu J, Goldberg MA, Bunn HF (1999) Inhibition of hypoxia-inducible factor 1 activation by carbon monoxide and nitric oxide. Implications for oxygen sensing and signaling. J Biol Chem 274: 9038-9044.
-
Hagen T, Taylor CT, Lam F, Moncada S (2003) Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 302: 1975-1978.
-
Callapina M, Zhou J, Schmid T, Köhl R, Brüne B (2005) NO restores HIF-1alpha hydroxylation during hypoxia: role of reactive oxygen species. Free Radic Biol Med 39: 925-936.
-
Yasuda H, Nakayama K, Watanabe M, Suzuki S, Fuji H, et al. (2006) Nitroglycerin treatment may enhance chemosensitivity to docetaxel and carboplatin in patients with lung adenocarcinoma. Clin Cancer Res 12: 6748-6757.
-
Yasuda H (2008) Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide 19:205-216.
-
Arrieta O, Blake M, de la Mata-Moya MD, Corona F, Turcott J, et al. (2014) Phase II study. Concurrent chemotherapy and radiotherapy with nitroglycerin in locally advanced non-small cell lung cancer. Radiother Oncol 111: 311-315.
-
Osaki T, Tatemoto Y, Yoneda K, Yamamoto T (1994) Tumorigenicity of cell lines established from oral squamous cell carcinoma and its metastatic lymph nodes. Eur J Cancer B Oral Oncol 30B: 296-301.
-
Pervin S, Singh R, Chaudhuri G (2001) Nitric oxide-induced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc Natl Acad Sci U S A 98: 3583-3588.
-
Cook T, Wang Z, Alber S, Liu K, Watkins SC, et al. (2004) Nitric oxide and ionizing radiation synergistically promote apoptosis and growth inhibition of cancer by activating p53. Cancer Res 64: 8015-8021.
-
Riganti C, Miraglia E, Viarisio D, Costamagna C, Pescarmona G, et al. (2005) Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res 65: 516-525.
-
Takabuchi S, Hirota K, Nishi K, Oda S, Oda T, et al. (2004) The inhibitory effect of sodium nitroprusside on HIF-1 activation is not dependent on nitric oxide-soluble guanylyl cyclase pathway. Biochem Biophys Res Commun 324: 417-423.
-
Olson N, van der Vliet A (2011) Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 25: 125-137.
-
Berchner-Pfannschmidt U1, Tug S, Kirsch M, Fandrey J (2010) Oxygen-sensing under the influence of nitric oxide. Cell Signal 22: 349-356.
-
Brüne B, Zhou J (2007) Nitric oxide and superoxide: interference with hypoxic signaling. Cardiovasc Res 75: 275-282.
-
Kasuno K, Takabuchi S, Fukuda K, Kizaka-Kondoh S, Yodoi J, et al. (2004) Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J Biol Chem 279: 2550-2558.
-
Taylor CT, Moncada S (2010) Nitric oxide, cytochrome C oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol 30: 643-647.
-
Callapina M, Zhou J, Schnitzer S, Metzen E, Lohr C, et al. (2005) Nitric oxide reverses desferrioxamine- and hypoxia-evoked HIF-1alpha accumulation--implications for prolyl hydroxylase activity and iron. Exp Cell Res 306: 274-284.
-
Kozhukhar AV, Yasinska IM, Sumbayev VV (2006) Nitric oxide inhibits HIF-1alpha protein accumulation under hypoxic conditions: implication of 2-oxoglutarate and iron. Biochimie 88: 411-418.
-
Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M (2006) Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 160: 1-40.
-
Ozben T (2007) Oxidative stress and apoptosis: impact on cancer therapy. J Pharm Sci 96: 2181-2196.
-
Conklin KA (2004) Chemotherapy-associated oxidative stress: impact on chemotherapeutic effectiveness. Integr Cancer Ther 3: 294-300.
-
Lamson DW, Brignall MS (1999) Antioxidants in cancer therapy; their actions and interactions with oncologic therapies. Altern Med Rev 4: 304-329.
-
Sasabe E, Yang Z, Ohno S, Yamamoto T (2010) Reactive oxygen species produced by the knockdown of manganese-superoxide dismutase up-regulate hypoxia-inducible factor-1alpha expression in oral squamous cell carcinoma cells. Free Radic Biol Med 48: 1321-1329.
-
Qing G, Simon MC (2009) Hypoxia inducible factor-2alpha: a critical mediator of aggressive tumor phenotypes. Curr Opin Genet Dev 19: 60-66.
