Adamantiades-Behçet's disease, Polymyositis, Muscle biopsy, Neurobehçet, Pain
Although Adamantiades-Behçet's disease (ABD) has a worldwide distribution, it is considered rare in the Americas, with a prevalence of 0.12-0.33:100.000 in the United States. The characteristic triad of recurrent oral aphthous lesions, genital ulcers and iridociclitis occurring more often in young adults during their third or fourth decade of life was initially described by [1,2] Hippocrates [3] but gained it's classic eponym to acknowledge the Greek ophthalmologist from Asia Minor (nowadays part of Asian Turkey) Benediktos Adamantiades and Turkish dermatologist Hulusi Behçet, who in first identified the symptomatic triad as an unique and distinct clinical entity. [4,5]
Since the first descriptions, the clinical spectrum of ABD has broadened [5-8] and today is considered a multisystem disorder, with a chronic course punctuated by recurrent acute exacerbations. Etiology remains unknown, [9] but genetic factors and autoimmune mechanisms have been reported with description of familial clusters and the association with the HLA-B51 antigen [10] with evidence of impaired cellular and humoral immunity [11]. From a histological point of view, ABD is characterized by leukocytoclastic or lymphocytic vasculitis associated with abnormal inflammatory tissue reaction [7,12].
The clinical picture of ABD affects nearly every organ in addition to [2] the classic triad, including manifestations in ocular, cutaneous, articular, vascular and gastrointestinal systems. Neurological manifestations, found in 10 to 30% of the patients, are recognized in the medical literature as Neurobehçet (NB) and include migraine-like headache, recurrent aseptic meningoencephalitis, encephalopathy, dementia, seizures, cranial nerve and bulbar palsy, pyramidal and extrapyramidal abnormalities, cerebellar ataxia, myelopathy, neurosensorial hearing loss, stroke, psychiatric disturbances, and a multiple sclerosis-like picture [6,9,13,14].
Although this broad spectrum of manifestations is widely recognized, myositis is rare. Here, we report the diagnostic challenge of a patient with ABD where polymyositis was the first and most important clinical manifestation.
A 55-year-old man was admitted to the University Hospital in Curitiba, Brazil, with a two-year history of progressive generalized weakness with proximal and lower limb predominance. He also complained of diffuse muscle soreness, weight loss, recurrent painful oral ulcers and painful swollen knee joints. Over this time, symptoms were continuous and added by hearing loss (mainly left sided), dysphagia and, during the last 3 months, tremor in his right arm. Past medical history was relevant for high blood pressure for the past 10 years treated with furosemide and amlodipine. There was no history of use of tobacco, alcohol, recreational drugs or risky sexual behavior. Family history was negative for a similar disorder.
Skin examination showed mild paleness with telangectasias in the malar and nasal areas. Pathergy test was positive. Tongue and internal lower lips presented multiple painful flat aphthous ulcers. Dorsal kyphosis was evident, associated with pronounced lumbar scoliosis. Right knee showed inflammatory signs and palpation of the ipsilateral thigh muscles was painful. Muscles were generally hypotrophic with 4 out 5 weaknesses throughout, decreased tone, and clear proximal predominance sparing the neck and face. Deep tendon reflexes were hyperreflexia with bilateral Hoffmann and right Babinski sign. Cranial nerves examination was only remarkable for bilateral neurosensorial hearing loss, more prominent on the left.
(Normal values in parenthesis) showed elevated CK 334 IU/l (10-160), aldolase 9.5 IU/l (1.2-7), lactate dehydrogenase 764 IU/l (150-480), erythrocyte sedimentation rate 66 mm (0-20), creatinine 3.7 mg/dl (0.7 to 1.3), uric acid 11.6 mg/dl (3.6-7.7), and 24-hour urine protein 8690 mg (40-80). Normal or negative results were found for the following: Cerebrospinal fluid analysis (chemistry, cytological, Gram staining, basic myelinic protein, electrophoresis), serum proteins immunoelectrophoresis, alpha-fetoprotein, carcinoembryonic antigen, liver and thyroid function, coagulation tests, screening tests for collagen vascular disease (LE cell preparation, rheumatoid factor, circulating immune complexes, anti-dsDNA, anti-nuclear and anti-Sm antibodies), serology for syphilis, HIV, HTLV 1-2, Hepatitis B and C. Electromyography (EMG): Proximal myopathic features on all limbs, with normal nerve conduction studies. Somatosensory evoked potentials: Normal.
Abdominal computed tomography was normal. Cervical magnetic resonance imaging (MRI): Signs suggestive of spondylosis with osteophytes and foraminal stenosis in C5-C6 and C6-C7. Brain MRI findings are shown in Figure 1A and Figure 1B.
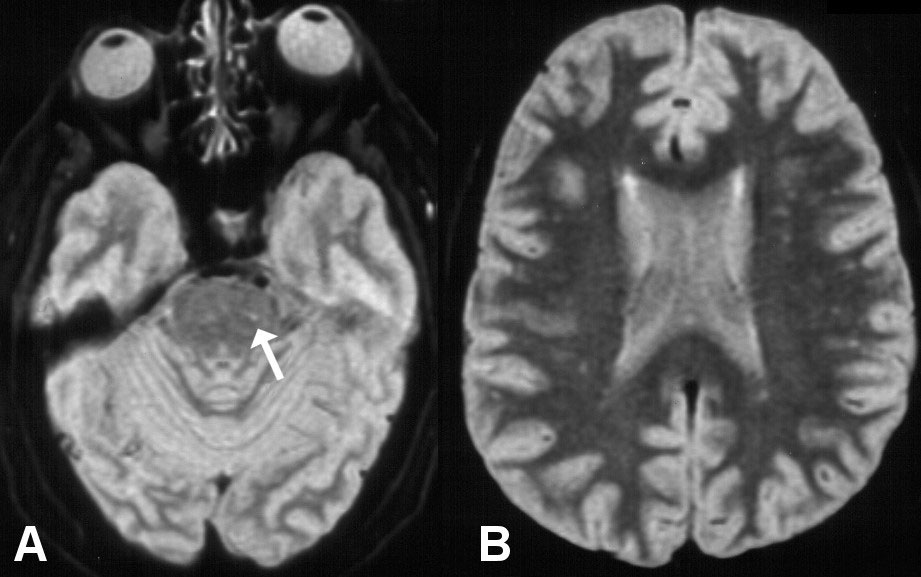 Figure 1: MRI axial T2-weighted A) showing hyperintense signal in the left antero-lateral pons; B) Showing diffuse hyperintense signal in the white matter consistent with ischemic small vessel disease.
View Figure 1
Figure 1: MRI axial T2-weighted A) showing hyperintense signal in the left antero-lateral pons; B) Showing diffuse hyperintense signal in the white matter consistent with ischemic small vessel disease.
View Figure 1
Skin and muscle biopsies are shown in (Figure 2A and Figure 2B). Audiometry: Moderate to severe bilateral neurosensorial hearing loss.
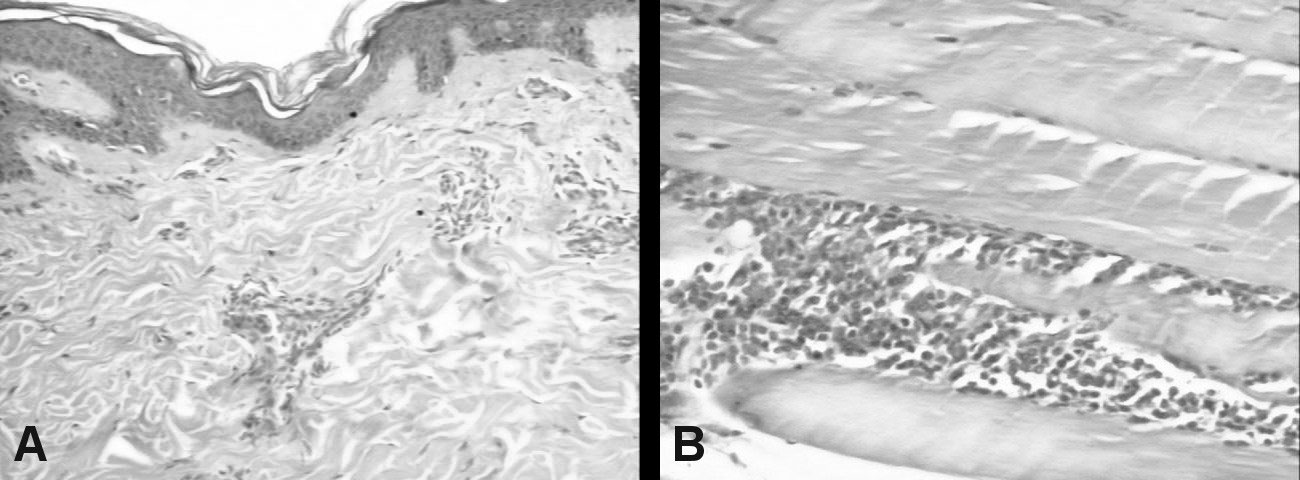 Figure 2: A) Skin biopsy (HE × 100): Mild epidermal lamelar hyperkeratosis. Superficial dermis showing moderate perivascular lymphocytic infliltration; B) Muscle biopsy (HE × 100): Mature adipose tissue, no atypical cells; Skeletal muscle fibers with moderate perimysial lymphocytic infiltrate.
View Figure 2
Figure 2: A) Skin biopsy (HE × 100): Mild epidermal lamelar hyperkeratosis. Superficial dermis showing moderate perivascular lymphocytic infliltration; B) Muscle biopsy (HE × 100): Mature adipose tissue, no atypical cells; Skeletal muscle fibers with moderate perimysial lymphocytic infiltrate.
View Figure 2
The neurological manifestations of ABD were described for the first time in 1941 by Knapp, and in 1954 Cavara and D'Ermo coined the term NB [15]. Given the lack of clinical and paraclinical pathognomonic markers, ABD [9] diagnosis is based on clinical criteria proposed by the International Study Group for Behçet Disease, which requires the presence of recurrent oral ulcers, in addition to two or more of the following: Recurrent genital ulcerations, inflammatory lesions of the eyes, specific skin lesions and positive cutaneous hyperreactivity test (pathergy) [15].
The case presented here fulfilled these criteria with recurrent oral ulcers, cutaneous vasculitis, central nervous system (CNS) lesions plus peripheral nervous system signs, arthritis, pericarditis, screening tests suggestive for glomerulonephritis, and positive pathergy test. The diagnosis of polymyositis (PM) was made on the basis of progressive proximal symmetric muscle weakness, elevated serum levels of skeletal muscle enzymes, suggestive EMG findings and diagnostic muscle biopsy. The main other causes of PM were excluded. Idiopathic PM is not characteristically accompanied by CNS vasculitis, arthritis and affects predominantly the cervical muscles. The screening tests for other collagen vascular diseases were normal or negative.
In fact, the first and most prominent feature of this case was the generalized muscle involvement with lower limb predominance. The histopathological examination showed vascular inflammation and lymphocytic infiltrate, which are considered characteristic features of this disorder [4,7].
In summary, ABD must be considered as a differential diagnosis of localized or generalized inflammatory muscle disorders especially when findings of multiple tissue and organ lesions or any symptom of the characteristic triad are present. Given the polymorphous and multisystemic nature of the syndrome, it's not surprising the report of "unusual" manifestations that will further expand the wide spectrum of NB.
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
None of the authors has any conflict of interest to disclose.