Ubiquitin proteasome pathway (UPP) failure has been reported in case of rabbit Limbal Stem Cell Deficiency (LSCD). In the present study, we examined the effect of LSCD on autophagy in comparison to LSCD's effect on UPP.
Rabbits with surgically induced LSCD were used. Corneal epithelial cells (CEC) were collected to measure the levels of autophagy and UPP biomarkers. Rabbit oral epithelial cells were isolated, cultured and treated with autophagy and proteasome inhibitors.
Immunofluorescent staining showed that both pathways were positive in normal corneal epithelium. However, while proteasome subunits were decreased in LSCD-CEC, autophagy biomarker MAPLC3B and ATG12-ATG5 complex were significantly increased in LSCD-CEC, compared to healthy CEC. These results indicate that LSCD-induced impairment of UPP may cause a compensatory stimulation of autophagy. However, despite autophagy up regulation, damaged and unwanted proteins, such as modified keratin K4 and K13 aggregates, still deposited and accumulated in LSCD-CEC without clearance, possibly contributing to CEC haziness. Proteasome inhibition in cultured oral epithelial cell sheet caused an increase in the expression of MAPLC3B and ATG12-ATG5 complex, supporting the observation that when UPP is failing, autophagy is stimulated. When autophagy was inhibited with chloroquine, proteasome activity was significantly increased compared to untreated cells. In addition, there a slight decrease in unmodified K4 and K13 expression levels with no keratin high molecular weight deposition when autophagy was inhibited.
We report that inhibition of autophagy leads to an activation in proteasome activity, which may stimulate protein degradation to alleviate the symptoms of LSCD.
Proteasome activation, Autophagy inhibition, Corneal epithelium
Protein homeostasis (proteostasis) plays an important role in stem cell identity and function, as it is central for self-renewal, pluripotency and cell fate decisions. To maintain proteostasis, protein degradation pathways operate in a balanced coordination for vital cellular functioning [1], prevent the passage of altered proteins to daughter during the asymmetric divisions, and consequently prevent an alteration of the resulting lineage of cells [2,3]. These proteolytic pathways are responsible for quality control of proteins and organelles, as well as for clearing misfolded, aged, malfunctioning and damaged intracellular components. Proteasomes and lysosomes are the two major proteolytic pathways by which cells degrade proteins for turnover and recycling purposes. Drs. Hershko, Ciechanover, and Rose were awarded the Nobel Prize in Chemistry in 2004 for the discovery of ubiquitin-mediated protein degradation. At that time, the ubiquitin proteasome pathway was characterized as the "Kiss of Death" because protein substrates are tagged with ubiquitin by the ubiquitination system prior to translocation inside the proteasome catalytic chamber for degradation by beta subunits [4]. Since then, there was an explosion of publications investigating the cellular function of proteasome, and, consequently proteasome inhibitors came to the market to treat cancer and suppress tumor formation [5]. Velcade, or Bortezomib/PS341, was the first therapeutic proteasome inhibitor to be tested in humans for its activity towards hematologic malignancies. Accelerated approval by the FDA was granted to the proteasome inhibitor Carfilzomib in 2012 for the treatment of patients with multiple myeloma. Several second-generation proteasome inhibitors are currently in clinical trials for the treatment of hematologic malignancies and solid tumors [6].
Lysosomes are membrane-enclosed cytoplasmic organelles responsible for the degradation of autophagy-mediated delivery of altered macromolecules, protein aggregates, and invading pathogens. Lysosome is the catalytic component of the autophagy system. All macromolecules tagged via autophagy system are consequently digested by the lysosome [7]. Dr. Ohsumi was awarded in 2016 the Nobel Prize in Physiology or Medicine for discovering and elucidating mechanisms underlying autophagy [8].
In both proteasome and lysosome pathways, protein degradation is achieved in a confined compartment, the lumen of the lysosome and the catalytic chamber of the proteasome, which prevents any unspecific digestion. Both pathways are essential for quality control mechanisms and operate with high fidelity in cells. The role of proteasomes in neurodegenerative disorders [9], cardiovascular diseases [10] and liver diseases [11] is well documented. However, studies on the molecular composition, function, or regulation of proteasome complexes of the eyes are scarce. Proteasome subtype and plasticity in corneal epithelial cells is also not well documented. Proteasome activity has been reported to be vital to maintain and regulate the levels of transcriptional factors and epithelial stem cell markers, such as DeltaNp63 [12] and PAX6 [13]. These two transcription factors are expressed in the limbal stem cells, as well as in corneal epithelial basal cells, which are progeny of limbal stem cells. Autophagy has also been reported to play a central role in limbal stem cell homeostasis and proliferation [14]. Both, proteasome and autophagy are particularly important for corneal epithelial cell (CEC) clearance and are the two most important pathways in clearing damaged intracellular proteins. As failure of UPP was previously reported [15], we examined in the present study the effect of limbal stem cell deficiency (LSCD) on autophagy in comparison to LSCD's effect on ubiquitin proteasome pathway (UPP). Hence, our aim is to understand the molecular mechanism(s) clearance of damaged intracellular proteins in corneal epithelial cells with LSCD. Understanding the complexity of proteasome and autophagy pathways and their explicit interactions in corneal tissue will provide the foundation for the development of new therapeutic strategies for LSCD.
New Zealand white rabbits weighing 2.5-3 kg were used. They were maintained according to the Guidelines of Animal Care, as described by the National Academy of Sciences published by the Institute of Laboratory Animal Resources Commission on Life Sciences National Research Council. The experimental protocol was approved by the IACUC and performed as previously reported [16]. Briefly, rabbits were sedated, subjected to lamellar limbectomy and followed for 3 months until LSCD was confirmed stable.
Corneal epithelial cells (CECs) were collected as previously reported [17]. Briefly, healthy (control) and diseased (LSCD) rabbit corneal epithelial cells were collected by exposing the corneas to 20% isopropyl alcohol. The corneas were then washed 3 times with saline and epithelial cells were scraped and removed from the corneal surface using a scalpel blade. The collected cells were then suspended in lysis buffer containing 20mM Tris-HCl pH = 7.5, glycerol 10% EGTA 1mM, DTT 1mM, protease and phosphatases inhibitor cocktail, as instructed by the supplier (Sigma, St Louis, MO). Cell membranes were disrupted by pipetting 25 times the cells trough a hypodermic 18G needle.
Paraffin-embedded rabbit corneal tissue sections were used to conduct immunohistochemical analysis. Slides were stained in immunofluorescent staining using MAPLC3B (Santa Cruz Inc., Santa Cruz CA), ATG12-ATG5 complex (Novus Biological, Centennial, CO), constitutive proteasome (CPR) beta1 (Enzo Life Sciences, Inc.) antibodies. Alexa Fluor® 488 and Alexa Fluo® 568 donkey anti-rabbit/mouse/goat fluorophore conjugated secondary antibodies were used. Staining cell nuclei was performed using propidium iodide (Invitrogen, Eugene, OR). Stained tissue sections were analyzed using a Nikon 400 fluorescent microscope. Picture processing and analysis was performed using Adobe Photoshop CS5.
Rabbit's buccal mucosa was biopsied, and tissue was cut in pieces and placed in Dispase (Millipore Sigma St. Louis, MO), solution to dissociate the tissue and isolate epithelial cells [16]. Epithelial cells were then cultured as previously reported [16].
Briefly, cells were grown in co-culture with mouse NIH3T3 feeder on TransWell insert (Corning Inc. St. Louis, MO). After two weeks of cell culture, proteasome inhibitor (50nM) and chloroquine (20mM) were separately added to cell culture media and cells were incubated for 24 hours before collection. Cells were then washed and scrapped of in lysis buffer for enzyme assays and biochemical proteomic analysis.
Cell culture experiments were carried out with one rabbit biopsy. Cells were isolated and cultured in three separate wells to obtain the n = 3 replicates. It is not possible to perform three biopsies and three-cell isolation in the same day. Experiments were reproduced another time with another rabbit biopsy to investigate the reproducibility among biological replicates.
One µg of total protein from cell lysates was used. Reaction mixture contained 50 mM Tris–HCl pH = 8.1 mM DTT, and 40 µM Suc-Leu-Leu-Val-Tyr-AMC (Sigma Aldrich, St. Louis, MO) substrate for chymotrypsin-like activity. The mixture was then incubated for 30 min at 37 ℃. The reaction was stopped by adding 100 µM monochloroacetate and 30mM sodium acetate pH=4.3. Fluorescence was determined by measuring the release of AMC (λ excitation: 370nm, λ emission: 410nm), using a Perkin Elmer LS 30 spectrofluorometer.
Two µg of total protein from sample homogenates were separated by SDS-PAGE gels and transferred to a PVDF membrane (Bio-Rad, Hercules, CA) for 1h in 25mM Tris-HC1 (pH = 8.3), 192mM Glycine and 20% methanol. Membranes were probed with primary antibodies against proteasome subunits beta 5, FK2 (used to detect high molecular weight poly-ubiquitinated proteins) from (Enzo Life Sciences, Inc.), MAPLC3B, and K13and K4 from (Santa Cruz Inc., Santa Cruz CA), Beta Actin (Sigma, St. Louis, MO), ATG12-ATG5 complex (Novus Biological, Centennial, CO), LAMP2 from (Abcam, Cambridge, MA), GAPDH (Millipore Burlington, MA). Polyclonal and monoclonal HRP-conjugated antibodies were used as secondary antibodies. Membranes were subjected to chemiluminescence detection using Luminal according to the manufacturer's instructions (Amersham Pharmacia Biotech, Piscataway, NJ).
Data were obtained from at least three different biological samples. Bars represent mean values ± SEM. P values were determined by one-way ANOVA and Student–Newman Keuls for multiple group comparisons (Sigma-Stat softdish, San Francisco, CA). Statistical significance was set at p= or < to 0.05. Bar graphs were shown as Mean+/- SEM, n = 3-5 biological replicates.
Limbal Stem Cell Deficiency (LSCD) has previously been found to be associated with proteasome dysfunction in corneal epithelial cells [15]. These results are confirmed in the present study by measuring the expression of proteasome subunits beta [8], as this proteasome subunit carries the major proteasome catalytic activity, the chymotrypsin-like activity. Figure 1A shows that proteasome subunit beta [5] levels were significantly decreased in LSCD-CEC, when compared to healthy control CEC, indicating a decreased proteasome activity in the ocular surface. However, autophagy's proteins microtubule-associated protein 1 light chain 3 B (MAPLC3B) (Figure 1B) and ATG12-ATG5 complex (Figure 1C) expressions were found elevated in LSCD-diseased CEC, when compared to control CEC, suggesting an activation of autophagy in the ocular surface with LSCD. We examined MAPLC3B and ATG12-ATG5 complex as they play an important role in autophagosome formation.
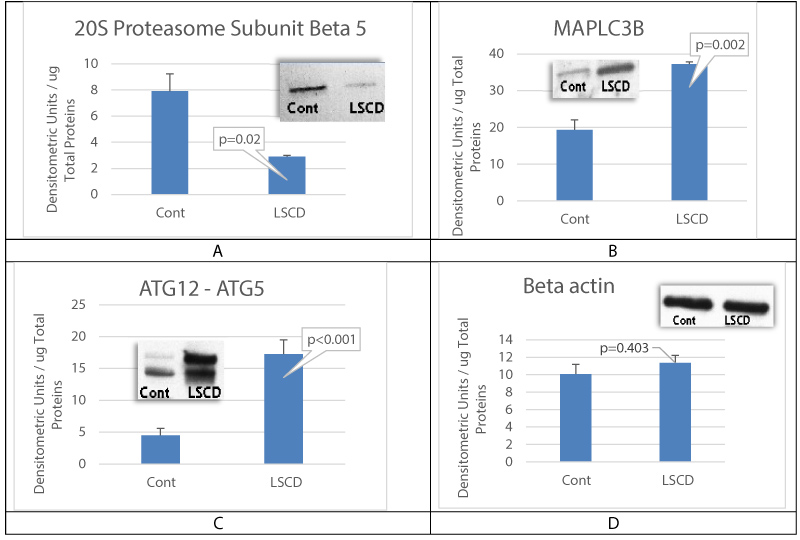 Figure 1: LSCD is associated with proteasome dysfunction and activation of autophagy in corneal epithelial cells.
Figure 1: LSCD is associated with proteasome dysfunction and activation of autophagy in corneal epithelial cells.
A is measurement of proteasome subunit beta 5 levels in rabbit healthy CEC (Control) and in LSCD-CEC (LSCD). B and C are respectively measurements of microtubule-associated protein 1 light chain 3 B (MAPLC3B) and ATG12-ATG5 complex levels in healthy rabbit CEC (Control) and in LSCD-diseased CEC (LSCD). D is the sample loading control with beta-actin staining. Mean +/- SEM, n = 3 biological replicates.
View Figure 1
Rabbit corneal tissue sections were used to morphologically evaluate the expression of proteasome and autophagy proteins. Figure 2 shows that proteasome beta 1 subunit was expressed in all corneal epithelium cells in control corneal tissue. The expression of proteasome beta 1 subunit was decreased in LSCD-diseased corneal epithelial cells (Figure 2A and Figure 2B). However, the expression of MAPLC3B and ATG12-ATG5 complex was scattered in healthy corneal epithelium and markedly expressed in LSCD-diseased corneal epithelium (Figure 2C, Figure 2D, Figure 2E and Figure 2F), respectively.
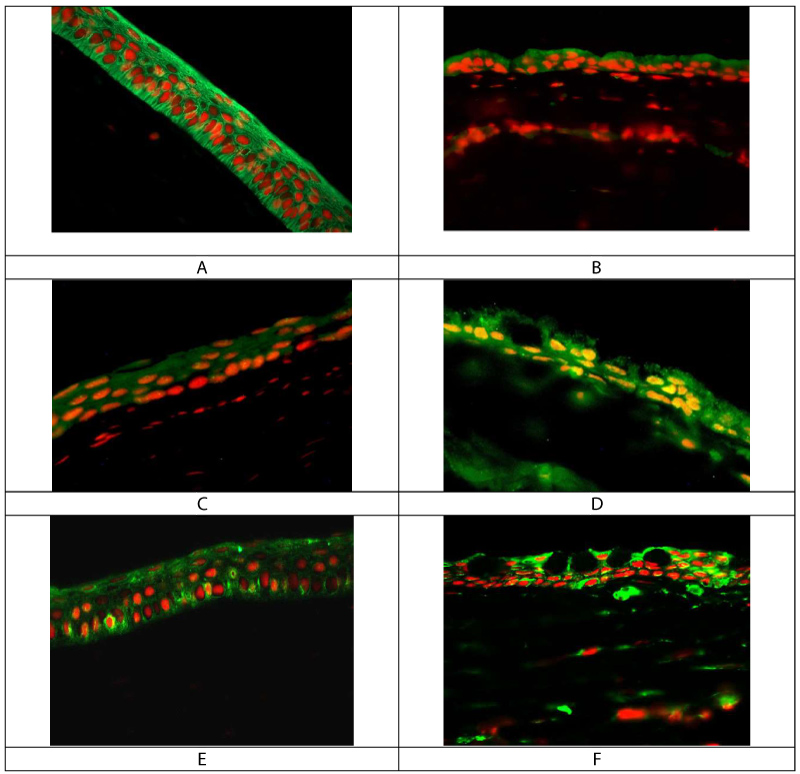 Figure 2: Immunofluorescent staining of rabbit corneal tissue sections.
Figure 2: Immunofluorescent staining of rabbit corneal tissue sections.
A and B are respectively control and LSCD corneal epithelium, stained with proteasome subunit beta 1. C and D are respectively control and LSCD corneal epithelium stained with MAPLC3B. E and F are respectively control and LSCD corneal epithelium stained with ATG12-ATG5 antibody that detect both proteins as a complex. Mag x40.
View Figure 2
Proteasome inhibition was achieved in cultured oral mucosal epithelial cell sheet for 24 hours before cell collection. MAPLC3B and ATG12-ATG5 complex expression levels were then measured using Western blot analysis. Proteasome chymotrypsin-like activity was significantly inhibited by proteasome inhibitor (Figure 3A). The expression of MAPLC3B was increased, as were the levels of autophagy conjugation factor ATG5 and ATG12 in cells treated with proteasome inhibitor (Figure 3B and Figure C).
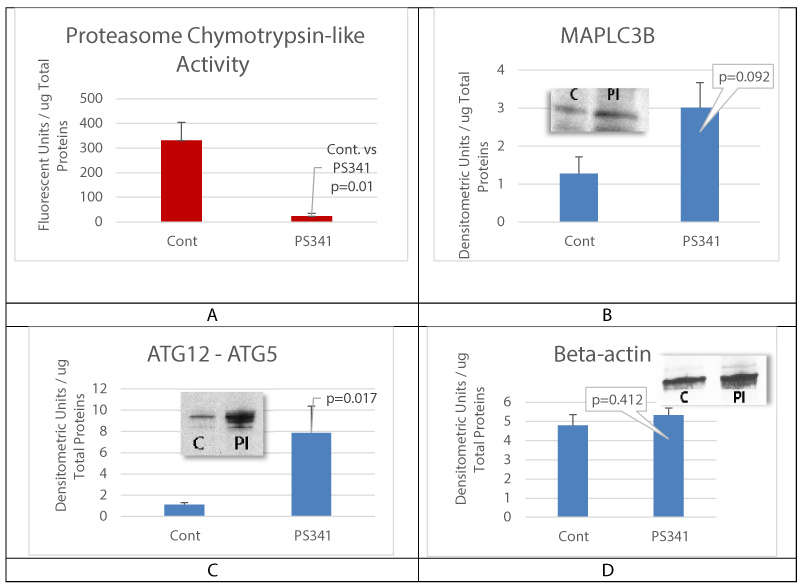 Figure 3: Proteasome inhibition activates autophagy.
Figure 3: Proteasome inhibition activates autophagy.
A is proteasome chymotrypsin-like activity assay; B and C are respectively MAPLC3B and ATG12-ATG5 complex levels in cultured oral mucosal epithelial cell sheet (OMECS) treated with proteasome inhibitor (PI). Control is untreated OMECS. Note that proteasome inhibition (PI) lead to a significant increase in ATG12-ATG5 complex expression levels. D is beta-actin levels measurement for gel loading control. Mean+/- SEM, n = 3 biological replicates.
View Figure 3
Autophagy inhibition was achieved by incubating cultured oral mucosal epithelial cell sheet with Chloroquine (ChQ) at 2mM for 24 hours before cells lysis and biochemical analysis. Proteasome chymotrypsin-like activity was measured, and results showed a significant increase in proteasome activity (Figure 4A), suggesting that autophagy inhibition may lead to proteasome activation. The expression of proteasome subunit beta 5, the active site of proteasome Chymotrypsin-like activity, was unchanged (Figure 4B). It is possible that the mechanism of proteasome activation does not required an increase in beta 5 expression but rather an increase in the proteasome subtype assembly. The levels of MAPLC3B and ATG12-ATG5 complex were also unchanged (Figure 4B, Figure 4C and Figure 4D). Lysosome-associated membrane protein 2 (LAMP2) was found up regulated with ChQ treatment (Figure 4E), which documents efficient autophagy inhibition in our experiment.
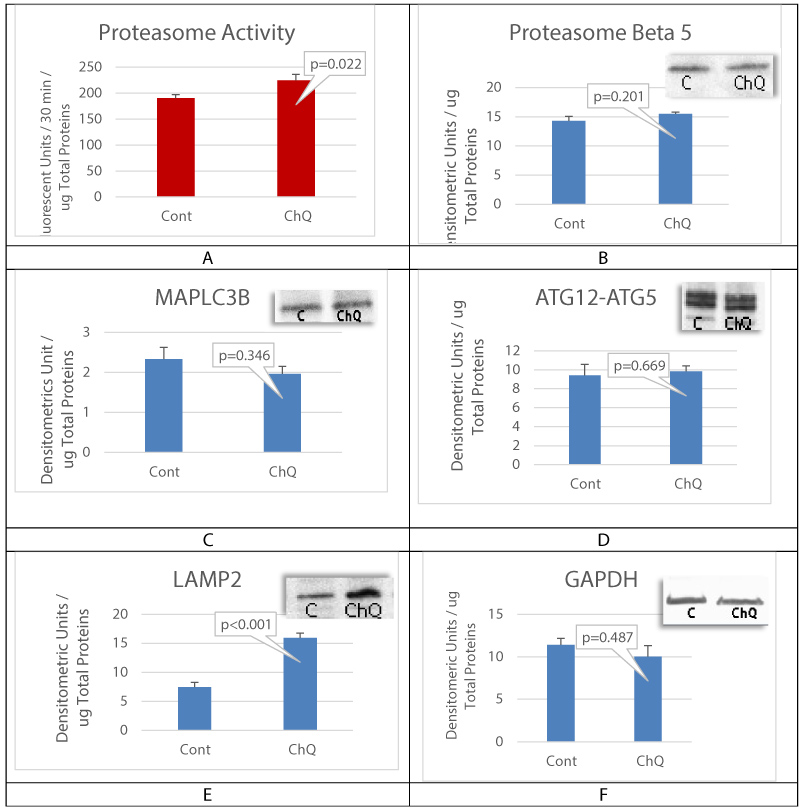 Figure 4: Autophagy inhibition activates proteasome.
Figure 4: Autophagy inhibition activates proteasome.
A is proteasome chymotrypsin-like activity assay; B is proteasome subunit expression levels. C and D are respectively autophagy component MAPLC3B and ATG12-ATG5 complex levels. E is LAMP2 expression levels and F is GAPDH levels verifying the samples gel loading control. Mean+/- SEM, n = 3 biological replicates.
View Figure 4
Results showed an increase in the expression of autophagy biomarkers when proteasome activity was decreased in corneal epithelial cells with LSCD. Using cell culture, the mechanistic in vitro experiments also demonstrated that proteasome inhibition is associated with an activation of autophagy. However, despite the increase in the expression of autophagy biomarkers, damaged proteins and unwanted proteins were still accumulated without clearance (Figure 5A, Figure 5C, and Figure 5E). When autophagy was inhibited, there was no accumulation of damaged and aggregated proteins and results showed a slight reduction in the levels of cytokeratin K4 and K13 expression in cells treated with chloroquine, suggesting that autophagy inhibition may lead to reduction in keratins accumulation in case of proteasome failure. Modulating the activity of these two pathways is a promising approach to increase damaged protein clearance by the proteasome and improve corneal epithelial cell function Figure 6.
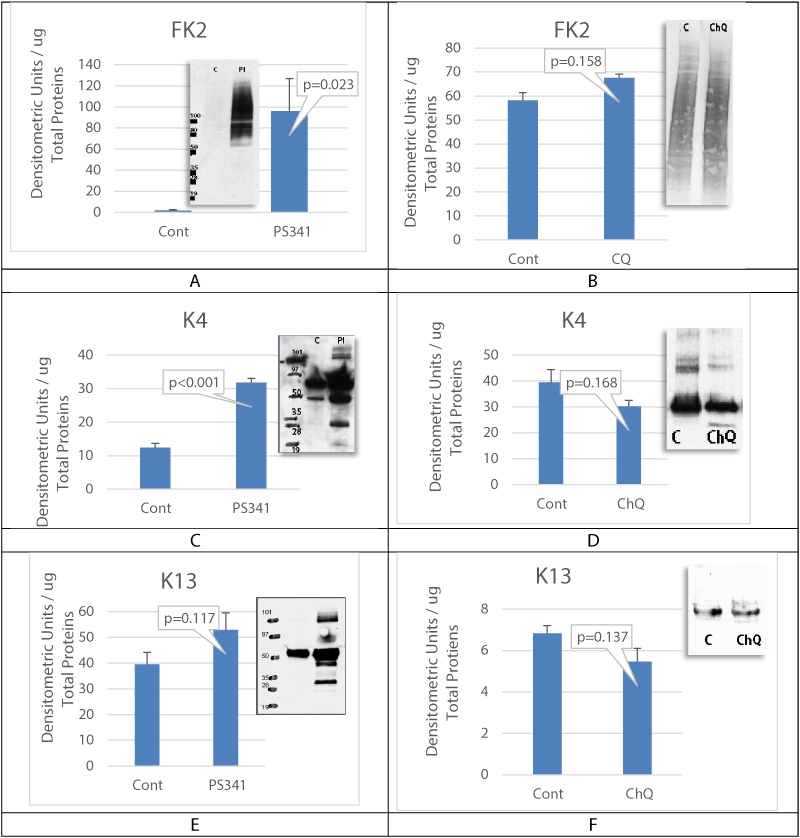 Figure 5: Autophagy inhibition activates proteasome.
Figure 5: Autophagy inhibition activates proteasome.
A, C and E are cultured cells treated with proteasome inhibitor. B, D and F are cultured cells treated with chloroquine to inhibit autophagy. FK2 antibody staining showed the accumulation of poly-ubiquitinated proteins in case of proteasome inhibition. The whole poly ubiquitin smear was measured. Keratins K4 and K13 (respectively 59 and 55 kDa bands) were significantly accumulated when proteasome was inhibited. Control is untreated OMECS. Mean+/- SEM, n = 3 biological replicates.
View Figure 5
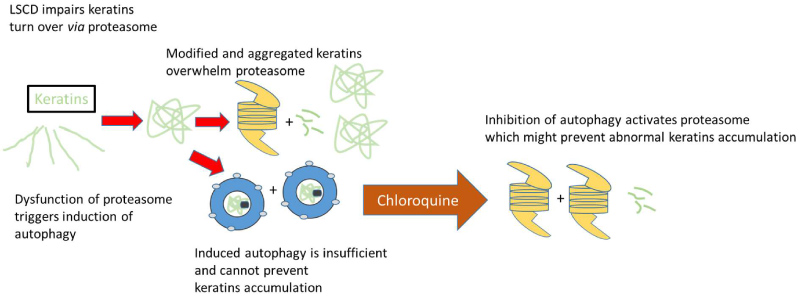 Figure 6: Schematic and hypothetical illustration of pathological cascade of events in corneal epithelium with LSCD.
Figure 6: Schematic and hypothetical illustration of pathological cascade of events in corneal epithelium with LSCD.
Accumulation of modified cytokeratin proteins (K13 and K4) may overwhelm the capacity of proteasomal degradation with accumulating protein burden leading to further impairment of these pathways. Autophagy is induced as a compensatory action, but is not sufficient to preserve protein homeostasis, which may promote cellular dysfunction in corneal tissue. Autophagy inhibition activates constitutive proteasomes and could improve corneal transparency.
View Figure 6
In summary, the present study demonstrates that in corneal epithelial cells with LSCD, proteasome dysfunction is associated with an activation of autophagy, which was not efficient in clearing abnormal and damaged accumulated proteins.
The present study shows evidence that protein degradation mechanisms are unbalanced, and consequently abnormal proteins accumulate to causing cellular dysfunction in case of LSCD. Constitutive proteasome (CPR) activity was found decreased in corneal epithelial cells with LSCD [15]. CPR is responsible for the major clearance and turnover of damaged and abnormal proteins in the cytosol. Proteins identified for degradation are first tagged with ubiquitin through multiple reactions of poly-ubiquitination and are finally digested inside the proteasome catalytic chamber formed by CPR [15,18].
Autophagy is a membrane trafficking mechanism in which cytoplasmic components, including organelles, are sequestered within double-membrane vesicles that deliver cytoplasmic bulk to the vacuole/lysosome for degradation and recycling [19].
The results of the present study provide evidence that autophagy is induced when CPR activity is reduced in corneal epithelial cells with LSCD. The correlation between both proteolytic pathways have been reported in numerous studies [20,21] and the adaptive activation of autophagy is consistently reported when proteasome activity is decreased [22]. We showed that MAPLC3B was significantly upregulated in LSCD-diseased corneal epithelial cells, indicating its adaptive role in the pathogenesis of LSCD. MAPLC3B protein is important for the induction of autophagy through elongation and expansion of the phagophore to form the autophagosome [23]. Autophagy has two ubiquitin-like conjugation systems, the MAPLC3B and Atg12 systems. In Atg12 conjugation system, ATG5 covalently bounds to ATG12 and conjugate functions as an important regulator for MAPLC3B lipidation [24] and autophagosome formation [25]. ATG5 then bounds to ATG16 and facilitates cytoplasmic proteins delivery to lysosomes for degradation [26]. Along with MAPLC3B, ATG12-ATG5-ATG16 complex elongates the phagophore membrane and dissociates when autophagosome formation is complete [27]. We found that the conjugated ATG5-ATG12 complex was up regulated in LSCD-diseased corneal epithelial cells.
To further understand this phenomenon, cultured oral mucosal epithelial cell sheet were treated with proteasome inhibitor. The levels of autophagy elements were then measured. In OMECS proteasome caused a significant upregulation of ubiquitin-like conjugation ATG12-ATG5 complex but did not significantly upregulate the levels of other ubiquitin-like conjugation complexes, i.e., MAPLC3B, in OMECS. Literature reports an increase in all ubiquitin-like systems of autophagy in case of treatment with the proteasome inhibitor PS341 (Bortezomib, Millennium Pharmaceuticals Inc.) [28]. In addition, early steps of autophagosome formation require ATG12 to be up regulated and covalently bound d to ATG5 [29]. LSCD caused a significant up regulation of MAPLC3B and ATG12-ATG5. It is possible that pharmacologic-induced proteasome inhibition will be different from LSCD-induced CPR dysfunction that was translated with a shift of proteasome population from CPR to IPR (immunoproteasome) due to high levels of inflammation [15]. It has been well documented that IPR is responsible for generating peptides for antigen presentation, but recent studies point toward the additional role of IPR, not only in activating cytokine productions, but also in the clearance of oxidized and altered protein [30-31]. The latest role of IPR in protein degradation and protein quality control, as well as the consequences of proteasome population shift in LSCD are still not well understood. Further studies will examine the effect of IPR activation in modulating autophagy and thus protein clearance.
A previous study demonstrated that proteasome system was affected and changed in case of LSCD [15]. However, despite autophagy up regulation, damaged and unwanted proteins, especially modified keratin K4 and K13 aggregates, were still deposited and accumulated in LSCD-diseased corneal epithelial cells without clearance [17], possibly contributing to corneal epithelial cell haziness.
In case of LSCD, the ocular surface is eroded and is clinically characterized by recurrent epithelial defects, conjunctivalization, neovascularization, and corneal opacity. Conjunctivalization is the hallmark of LSCD because the limbus is the barrier to conjunctival epithelial and goblet cells invasion of central cornea with or without neovascularization [32]. Conjunctival epithelium is a thin transparent layer of cells covering the sclera - the white part of the eye. Corneal invasion by this transparent layer may not be totally responsible for the ocular surface haziness associated with LSCD-induced conjunctivalization. It is possible that invasive conjunctival epithelial cells are different type of cells with different pattern of keratins. Healthy corneal epithelial cells express keratin K3/K12 [33] and the absence of K3 is used as a diagnostic tool for LSCD [34] as the expression of corneal keratins changes by expressing K4/K13 instead of K3/K12, which is typically the indication of corneal conjunctivalization [35,36]. In a previous study [17], the pattern of K4/K13 expression in diseased CEC was found different from those of healthy conjunctival epithelial cells [37]. Briefly, keratins modified protein isoforms were detected and high molecular levels of keratins formed, indicating the formation of corneal keratin aggregates (CKAGG). It is not known whether LSCD-induced formation of CKAGG is a contributing factor to corneal opacity.
The ubiquitin proteasome pathway (UPP) is responsible for degrading misfolded proteins but was unable to sufficiently clear K4 and K13 in this study. It is possible that UPP is overcrowded by the build-up of K4 and K13, leading to the corneal aggresome formation, which could activate autophagy pathway to clear the buildup of modified K4 and K13 in the ocular surface of rabbit with LSCD. Autophagy acts to clear large groups of proteins or even organelles by forming a membrane bound autophagosome, which fuses with a lysosome [38]. It is possible that autophagy was activated to clear modified K4 and K13 after UPP was overwhelmed but was still unable to prevent the accumulation of K4 and K13 in the corneal epithelial cells with LSCD.
We believe that the subsequent activation of UPP would effectively clear protein aggregates and restore ocular transparency. To find a method to improve the transparency of corneal epithelial cells with LSCD, we focused on modulating the activity of autophagy, which would then indirectly act to modulate the activity of UPP. When autophagy pathway was inhibited with chloroquine, proteasome chymotrypsin-like activity was significantly increased, compared to untreated cells. There was a trend of decreasing unmodified K4 and K13 levels with no keratin high molecular weight deposition when autophagy was inhibited. These results suggest that autophagy inhibition can increase the proteasome activity and possibly prevents accumulation of high molecular weight keratin deposition.
Grant support: Research is fully supported by Emmaus Medical Inc. Dr. Fawzia Bardag Gorce and Mrs. Kavita Narwani salaries were fully supported by Emmaus Medical Inc.; and Dr. Yutaka Niihara is the CEO and the Chairman of Emmaus Medical, Inc. Dr. Joan Oliva is an employee of Emmaus Medical, Inc. The rest of the authors has no support from Emmaus Medical, Inc. and no conflict of interest.