Cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD) are chronic pulmonary diseases that affect ~70,000 and 251 million individuals worldwide, respectively. Although these two diseases have distinctly different pathophysiologies, both cause chronic respiratory insufficiency that erodes quality of life and causes significant morbidity and eventually death. In both CF and COPD, the respiratory microbiome plays a major contributing role in disease progression and morbidity. Pulmonary pathogens can differ dramatically during various stages of each disease and frequently cause acute worsening of lung function due to disease exacerbation. Despite some similarities, outcome and timing/type of exacerbation can also be quite different between CF and COPD. Given these clinical distinctions, both patients and physicians should be aware of emerging therapeutic options currently being offered or in development for the treatment of lung infections in individuals with CF and COPD. Although interventions are available that prolong life and mitigate morbidity, neither disorder is curable. Both acute and chronic pulmonary infections contribute to an inexorable downward course and may trigger exacerbations, culminating in loss of lung function or respiratory failure. Knowledge of the pulmonary pathogens causing these infections, their clinical presentation, consequences, and management are, therefore, critical. In this review, we compare and contrast CF and COPD, including underlying causes, general outcomes, features of the lung microbiome, and potential treatment strategies.
Cystic fibrosis, Chronic obstructive pulmonary disease, Biofilms, Microbiology, Infection
CF is a lethal recessive autosomal disorder observed predominately among Caucasians [1]. The disease is caused by mutation of the cystic fibrosis transmembrane conductance regulator (CFTR), a protein expressed in many epithelia (Figure 1). By far the greatest negative consequence of CF is progressively deteriorating lung function. Loss of CFTR causes complications that also include pancreatitis, hepatic injury, nasal polyposis, digital clubbing, meconium ileus, and other intestinal obstructive symptoms [2]. An important activity of CFTR is to regulate anion (e.g. chloride (Cl-) and bicarbonate (HCO3-)) secretion and absorption in epithelial tissues [3]. Without this cellular activity, imbalance of exocrine secretion and composition leads to hyperviscous mucus in numerous secretory organs [4].
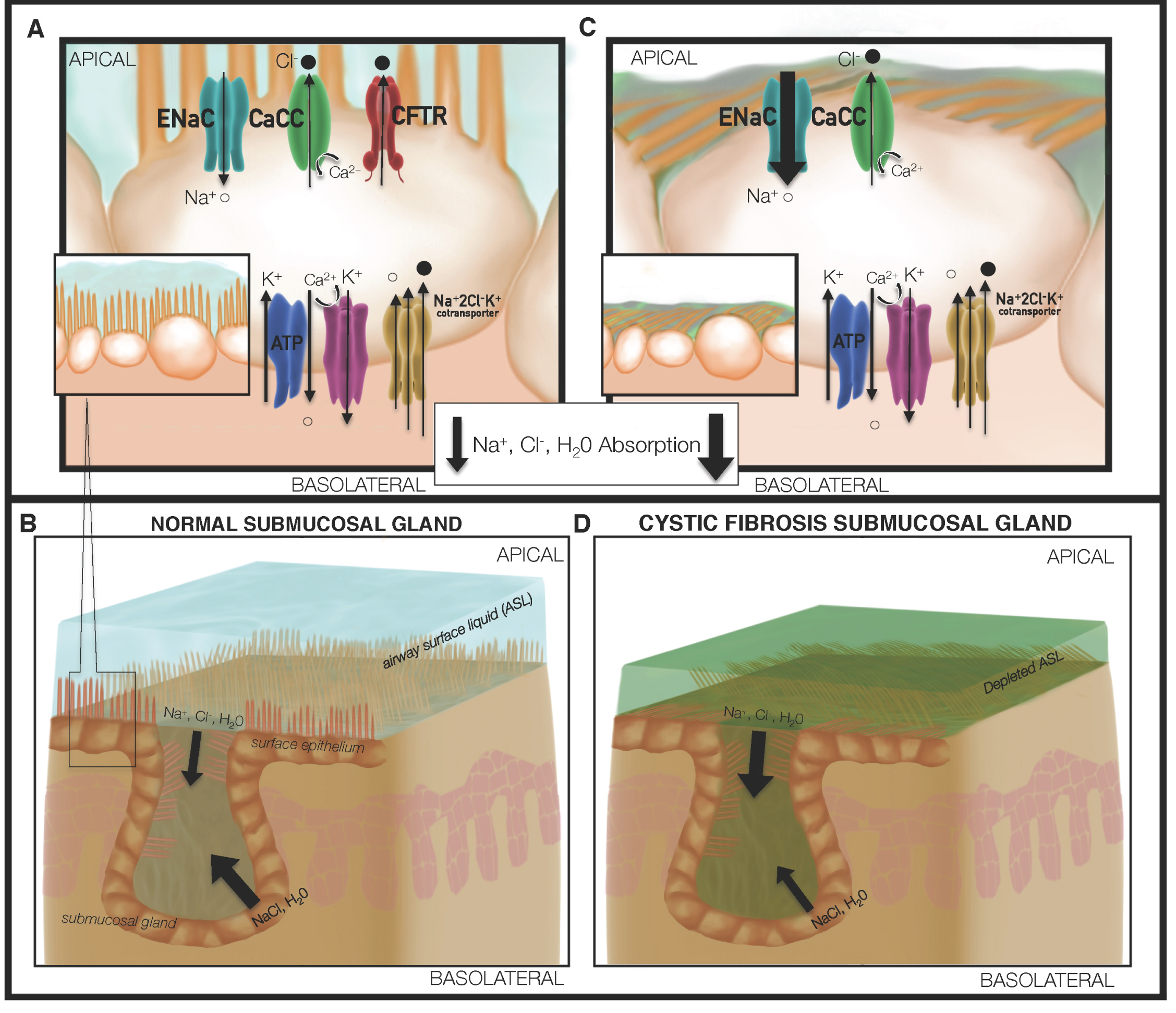 Figure 1: Loss of CFTR drives defective mucociliary transport in cystic fibrosis. (A) Normal mucus formation is dependent upon chloride, fluid, bicarbonate and pH homeostasis, which is in part maintained via CFTR activity; (B) The submucosal glands in lung tissues produce secretions that enable effective transport of mucus by ciliary beating of airway epithelial cells. In contrast, depletion of CFTR mediated fluid and electrolyte transport; (C) Alters periciliary fluid composition and is associated with enhanced potential difference attributable to ENaC mediated Na+ transport. The surface epithelium and submucosal gland abnormalities confer a surface liquid and mucus environment of lower pH, diminished surface liquid depth, and increasing mucous viscosity; (D) Combination of depleted ASL volume and fluidity, coupled with changes in ion balance, allow for the proliferation and reduced clearance of bacteria such as Pseudomonas aeruginosa.
View Figure 1
Figure 1: Loss of CFTR drives defective mucociliary transport in cystic fibrosis. (A) Normal mucus formation is dependent upon chloride, fluid, bicarbonate and pH homeostasis, which is in part maintained via CFTR activity; (B) The submucosal glands in lung tissues produce secretions that enable effective transport of mucus by ciliary beating of airway epithelial cells. In contrast, depletion of CFTR mediated fluid and electrolyte transport; (C) Alters periciliary fluid composition and is associated with enhanced potential difference attributable to ENaC mediated Na+ transport. The surface epithelium and submucosal gland abnormalities confer a surface liquid and mucus environment of lower pH, diminished surface liquid depth, and increasing mucous viscosity; (D) Combination of depleted ASL volume and fluidity, coupled with changes in ion balance, allow for the proliferation and reduced clearance of bacteria such as Pseudomonas aeruginosa.
View Figure 1
CF typically causes pulmonary symptoms that include chronic cough and sputum production, airway obstruction, wheezing and air trapping, and persistent colonization/infection by a myriad of microorganisms (6). Bacteria involved in such infections include Staphylococcus aureus, Pseudomonas aeruginosa, and Burkholderia cepacia. Others, such as Haemophilus influenzae, Klebsiella pneumoniae, Stenotrophomonas maltophilia, Achromobacter xylosoxidans, and non-tuberculous mycobacteria are increasingly recognized as important CF pathogens [5,6].
Nearly 2,000 mutations in CFTR have been documented, many of which are associated with clinical disease (CFTR2 database at cff.org). The most common mutation is Δ508. The phenylalanine at position 508 of the protein [7] is situated in the first CFTR nucleotide binding domain (NBD) and serves to both stabilize the overall NBD and allow proper interactions between NBD1 and downstream CFTR elements such as cytosolic loop 4 [8]. The Δ508 mutation leads to a protein that functions as an ion channel but is not trafficked efficiently to the cell surface and exhibits diminished half-life in the plasma membrane. The Δ508 protein also exhibits defective channel gating function [9]. Unlike normal airways, abnormalities of exocrine secretion lead to hyperviscous mucus, which adheres to the airway surface and apical membranes of numerous epithelia (an example of which is depicted in Figure 2). In airway glands, abnormalities prevent detachment of mucus strands from glandular ostea, compounding mucus viscosity and hindering clearance. Because mucociliary activity represents an essential component of airway defense against infection, the lungs become highly susceptible to bacterial colonization [10].
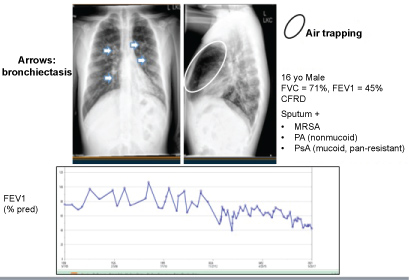 Figure 2: CF Chest X-ray and predicted FEV1 measurements over time.
View Figure 2
Figure 2: CF Chest X-ray and predicted FEV1 measurements over time.
View Figure 2
The lifespan of patients with CF has dramatically increased due to new therapeutic interventions and improved understanding of the disease (www.cff.org). In particular, median life expectancy in 1985 was < 25 years, and by 2017 had reached > 40 years. Despite these advances, morbidity and mortality attributable to respiratory infection remain prevalent. An enduring priority to increase the survival of individuals with CF has been the exploration of new and innovative antimicrobial, anti-infective, mucolytic, and other treatment strategies.
Chronic obstructive pulmonary disease (COPD) is an increasingly prevalent chronic disease affecting Americans and others worldwide, that is rapidly becoming a leading cause of death internationally [11,12]. However, unlike CF, COPD is a potentially preventable and non-lethal (with proper patient compliance) and a treatable disease that is usually characterized by airflow limitation, intensified lung and systemic inflammation, episodic exacerbations, and comorbidities [13]. The systemic nature of COPD is very different from that of CF. The manifestation of systemic COPD includes a variety of factors that are not limited to cardiovascular disease, osteoporosis, depression, weight loss and skeletal muscle issues [14]. Until recently, airflow limitation, a reduction in the exhalation of air due to increased airway resistance and dynamic airway collapse has been an essential element for the diagnosis of COPD. Airflow limitation is quantified by the ratio of FEV1 (forced expiratory volume in 1 sec) to FVC (forced vital capacity) and is present when this ratio is less than a threshold value. Typical threshold values are an absolute value of 0.7 or the lower limit of normal (LLN). The LLN represents the 5th percentile of the distribution of the FEV1/FVC ratio in nonsmokers with no lung disease. Recent investigations show that some individuals with radiographic evidence of emphysema may not have physiologic evidence of airflow limitation [15]. COPD is commonly viewed as two processes, chronic bronchitis and emphysema, with overlapping clinical, radiographic, and physiologic findings (please refer to Figure 3, Figure 4 and Figure 5 for examples).
 Figure 3: Normal posterior anterior chest radiograph. Vascular markings within respiratory parenchyma attenuate in the lateral one third of both lungs. The diaphragms have a dome shaped configuration.
View Figure 3
Figure 3: Normal posterior anterior chest radiograph. Vascular markings within respiratory parenchyma attenuate in the lateral one third of both lungs. The diaphragms have a dome shaped configuration.
View Figure 3
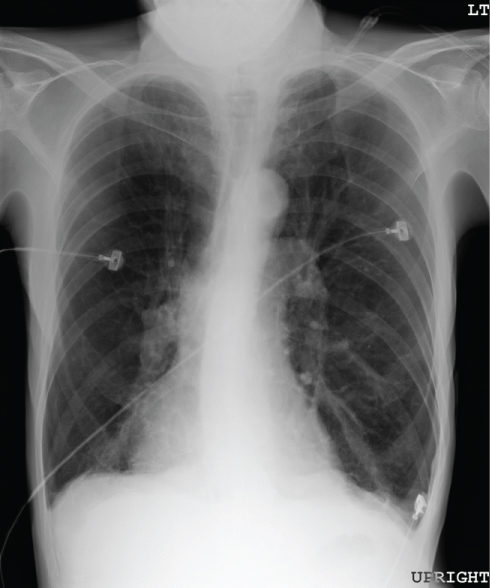 Figure 4: Chest radiograph of an individual with severe emphysema. Lung fields are greatly enlarged and exhibit paucity of vascular markings throughout. Diaphragms are flattened and have lost their normal dome configuration.
View Figure 4
Figure 4: Chest radiograph of an individual with severe emphysema. Lung fields are greatly enlarged and exhibit paucity of vascular markings throughout. Diaphragms are flattened and have lost their normal dome configuration.
View Figure 4
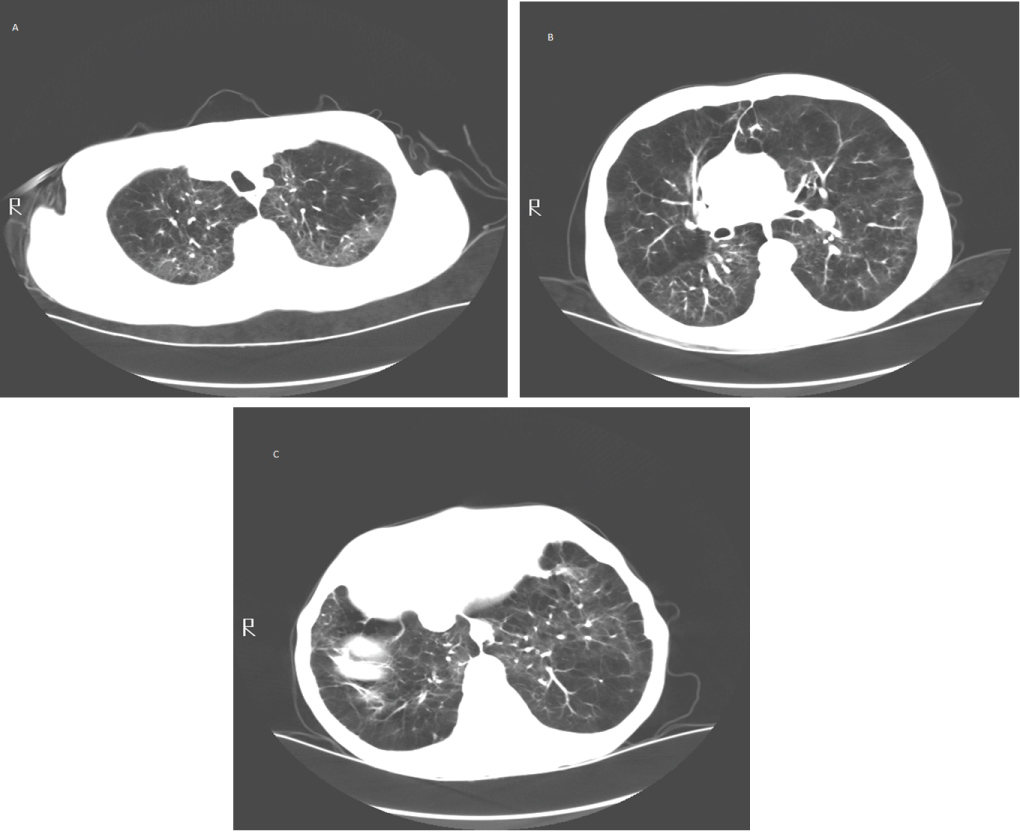 Figure 5: (A) Upper lung zone; (B) Mid lung zone; (C) Lower lung zone. Computed tomography of an individual with severe emphysema. Lung parenchyma is destroyed and replaced by cysts and blebs. Areas of more normal lung are compressed by hyperinflated emphysematous lung tissue.
View Figure 5
Figure 5: (A) Upper lung zone; (B) Mid lung zone; (C) Lower lung zone. Computed tomography of an individual with severe emphysema. Lung parenchyma is destroyed and replaced by cysts and blebs. Areas of more normal lung are compressed by hyperinflated emphysematous lung tissue.
View Figure 5
There may be a connection between COPD-bronchiectasis as a subtype of COPD, but this notion remains unclear. Thus, at this time, it is premature to classify COPD-bronchiectasis as a subtype of COPD and therefore comparison between CF and COPD-bronchiectasis lacks merit. The only paper that comes remotely close to such a posit is by Blasi, et al. [16]. Still, epidemiologic studies of COPD implicate multiple etiologic risk factors including allergies and hyper-responsiveness to airborne particulates, as well as recurrent bronchopulmonary infections [17]. But by far, the single most deleterious cause is long-term exposure to tobacco smoke. In 2011, the prevalence of COPD among US adults was 5.2% for men and 6.7% for women and 76% of those individuals with self-identified COPD were current or former smokers [18]. COPD normally presents later in life after prolonged risk factor exposure. Although tobacco smoke inhalation is the major risk factor for the development of COPD, a comparatively small proportion of smokers develop the condition. Nearly one quarter of individuals with COPD have no smoking history [19]. Indoor and outdoor air pollution, workplace aerosolized chemicals, dusts, and fumes, and respiratory infections including tuberculosis are other exposures associated with the development of COPD [19-21]. Genetic factors predisposing individuals to COPD are poorly understood.
The primary clinical manifestations of COPD are breathlessness, cough, and sputum production dyspnea [22]. Breathlessness is caused mainly by dynamic hyperinflation with exertion which produces air trapping and diminished tidal volumes. Airflow limitation may also contribute to shortness of breath. Cough and sputum production are caused by hyperplasia and hypertrophy of mucus producing airway epithelial cells, colonization of the lower airway by bacterial pathogens, and the eventual structural derangement of the airways leading to the development of bronchiectasis (permanent enlargement and structural alteration of the lower bronchi) [23].
COPD is primarily a pulmonary disorder but may also affect multiple other organ systems. Much of the initial morbidity associated with COPD is caused by cardiovascular and other manifestations believed to be mediated through systemic inflammation. Other associated clinical findings include musculoskeletal disorders (osteopenia and osteoporosis that culminate in fractures), diabetes, anemia of chronic inflammation, and psychological effects such as anxiety and depression due to chronic breathlessness, reduced physical function, and social isolation. Lung inflammation and its subsequent systemic dissemination in COPD has also been implicated as a contributor to coronary artery disease, congestive heart failure, obesity, endocrinologic dysfunction, osteoporosis, and other health issues [24].
Prevention of COPD through smoking avoidance or cessation is the most important element of COPD management. Once the disorder has been correctly diagnosed, management is both pharmacologic and nonpharmacologic. COPD is treatable but cannot be reversed due to chronic lung remodeling and permanent anatomic destruction.
COPD medication management begins with a short acting bronchodilator and then gradually escalates with the addition of long acting anticholinergics, long acting beta agonists, and finally inhaled corticosteroids. Antibiotics and systemic steroids are often used to treat acute COPD exacerbations due to infections. Selective phosphodiesterase inhibitors and macrolide antibiotics may be used for individuals with frequent exacerbations. Not adhering to treatments is associated with a 40% increased likelihood of hospitalization [25]. Nonpharmacologic therapies include supplemental oxygen, vaccinations, lung volume reduction surgery, lung transplantation, and pulmonary rehabilitation.
COPD exacerbations occur episodically throughout the disease course and are characterized by increased respiratory symptoms, especially breathlessness, cough, and sputum production, that are more pronounced than their usual day to day variation [26]. Similar to CF, patients with COPD exhibit chronic inflammation and hypersecretion of mucus that may become more pronounced during exacerbations [17]. Mucus overproduction is associated with mucosal gland hypertrophy, increased numbers of epithelial goblet cells, and damaged cilia. This excessive mucus and deranged mucociliary clearance mechanism may manifest clinically as increased and forceful coughing with excessive phlegm production and may contribute to bacterial colonization and infection of the lower airways that propagate a cycle of progressive inflammation and derangement of lung structure. Just as in CF, disease worsening is associated with colonization and invasion of respiratory tissue by bacterial pathogens.
The lungs of CF patients become chronically infected with a myriad (potentially > 100 different genera) of bacteria, leading to a poorer clinical prognosis [27]. Major pathogens include members of non-tubercle mycobacteria (Mycobacterium abscessus, M. avium, M. intracellulare, M. fortuitum, M. gordonae, M. kansasii), Staphylococcus aureus, Pseudomonas aeruginosa, Burkholderia cepacia complex (BCC, genomovar I (B. cepacia), II (B. multivorans), III (B. cenocepacia), IV (B. stabilis), V (B. vietnamiensis), VI (B. dolosa), VII (B. ambifaria), VIII (B. anthina), IX (B. pyrrocinia), Burkholderia gladioli and Burkholderia pseudomallei. Minor pathogens include Achromobacter xylosoxidans, Inquilinus limosus, Ralstonia sp., Pandoraea apista, Streptococcus pneumoniae, Stenotrophomonas maltophilia, Haemophilus influenzae and Bordetella bronchiseptica. In addition to the aforementioned bactéria, strict anaerobes have increasingly been encountered and include the genera, Prevotella, Veillonella, Propionibacterium and Actinomyces. These organisms were derived from a superb microbiological CF assessment by Coutinho, et al. [28].
With this information in hand, we must emphasize that the predominant infecting organisms change with age; for example, S. aureus is often a major species during childhood. As CF patients become older, the opportunistic pathogen P. aeruginosa becomes more firmly established, eventually outgrowing most other species, which remain present in lesser numbers [29,30].
Patients given antimicrobial treatment focused on P. aeruginosa eradication may develop niches that other pathogens occupy, leading to a remarkably complicated biologic flora [31]. S. aureus, and the more resilient methicillin-resistant S. aureus (MRSA), are Gram-positive cocci that are isolated from 71% and 26% of CF patients, respectively. The prevalence of these organisms has been increasing.
As described above, S. aureus is typically an early colonizer of CF lungs, likely because the organism is a common commensal on skin and in the respiratory tract of humans. Colonization occurs at an early age and is usually present at some level throughout the life of patients with the disease. Before standardized use of antibiotic regimens such as respiratory flucloxacillin and dicloxacillin [32], S. aureus was the leading cause of death in CF. This organism is now managed with more effective antimicrobials. It is important to note, however, that high staphylocidal activity for an antibiotic may not be sufficient to eradicate an infection, in part due to inability of cilia to properly expel viscous and infected mucus from the lungs of individuals with CF [33].
S. aureus isolates from individuals with CF have been shown to exhibit a distinct "small colony" morphology [34,35] (Figure 6). Colonies of this type have decreased virulence properties, produce less alpha toxin (a hemolytic protein), and elaborate no pigment [36]. This phenotype may help mask the organism from recognition by the immune system, thereby preventing clearance. In addition, small colony variants have increased antibiotic resistance properties, and exposure to aminoglycosides (e.g. gentamicin) promotes conversion to the phenotype. Even after selective pressure elicited by antibiotic treatment is removed, small colony morphology persists [34-37]. Antibiotic resistance in this setting may result from lower bacterial uptake of aminoglycosides and/or use of exogenous nucleic acids to combat the effect of antifolates [38,39].
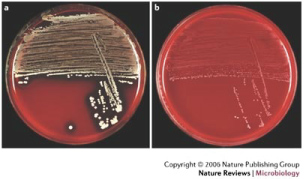 Figure 6: (A) Normal S. aureus; (B) S. aureus with small colony phenotype. With permission from [190]. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070213221.
View Figure 6
Figure 6: (A) Normal S. aureus; (B) S. aureus with small colony phenotype. With permission from [190]. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070213221.
View Figure 6
As CF patients become older, P. aeruginosa typically becomes more prominent despite aggressive antibiotic treatment [40,41]. Resistance is due in part to ability of the organism to form highly antibiotic- and phagocyte-refractory biofilms - complex microbial communities enmeshed within the thick CF airway mucus. The formation of biofilms is associated with the ability to quorum sense (QS), a process of inter-cellular signaling (i.e., bacterial communication) through secreted extracellular signaling molecules that coordinate biofilm formation and structure [42]. P. aeruginosa is known to use QS during CF lung infection, including QS autoinducers PAI-I and PAI-2, which are detected in CF sputum [43]. Bacteria in biofilms develop phenotypic distinctions compared with those bacteria associated with acute infections. For example, chronic organisms often become mucoid (alginate-overproducing), non-motile, non-flagellated, lipopolysaccharide-deficient, auxotrophic and/or antibiotic-resistant [44,45]. From among this multitude of alterations, the most common and clinically devastating is mutation of the mucA gene, encoding an anti-sigma factor that binds AlgT(U), a transcriptional regulator involved in production of alginate and the process of mucoid conversion. The mucoid phenotype is characterized by overproduction of highly viscous alginate expolysaccharide and represents an important step during establishment of chronic and fatal CF lung infections [46-49]. The switch or trigger for a mucoid phenotype has been associated with steep oxygen gradients within the thick airway CF mucus [50] (Figure 7).
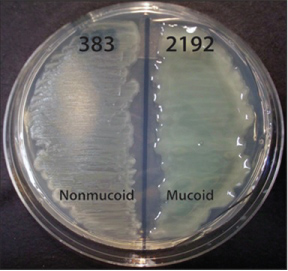 Figure 7: Phenotypes of two distinct P. aeruginosa CF isolates (383 and 2192) on L agar media. The strains were obtained 2 days apart from the same CF patient, and are otherwise isogenic [191]. The genome of strain 2192 has been sequenced [192], from [193].
View Figure 7
Figure 7: Phenotypes of two distinct P. aeruginosa CF isolates (383 and 2192) on L agar media. The strains were obtained 2 days apart from the same CF patient, and are otherwise isogenic [191]. The genome of strain 2192 has been sequenced [192], from [193].
View Figure 7
During chronic infection, high bacterial mutation rates facilitate P. aeruginosa adaptation to an ever-changing environment. "Hypermutable" strains are more common in CF lungs that are chronically infected compared with those that are acutely infected [51]. Such strains exhibit changes in proofreading and DNA repair, allowing for rapid development of strain to strain differences (potentially including mucA loss of function [52]. Polymorphisms observed in one patient sample may be restricted to that individual, suggesting genetic and phenotypic evolution occurs longitudinally after initial lung infection [51]. These individualized lung microbiomes can be attributed to the compartmentalized nature of the pulmonary anatomy and varying evolutionary pressures, such as differing concentrations of antibiotics that select for the more resistant organisms [53,54].
B. cepacia represents another clinically important pathogen in CF lung disease. This organism, formerly termed Pseudomonas cepacia, has been recategorized as B. cepacia complex (BCC), a group of at least 20 genetically distinct, but phenotypically similar bacteria [55-57]. Members of BCC are gram-negative, catalase positive, obligate aerobic bacilli that can persist in the presence of certain disinfectants and readily survive with minimal nutrition. Infection with BCC, first recognized in the CF patient population in the late 1970s, has been associated with severe worsening of CF pulmonary reserve and poor clinical prognosis [58-60]. Similar to P. aeruginosa, BCC has the ability to form biofilms in vivo, potentially impacting antibiotic resistance [61,62]. Although formation of BCC biofilms may help establish initial infection, in contrast to P. aeruginosa, there is an inverse relationship between exopolysaccharide production and decline of CF pulmonary reserve. This difference may be due in part to an increased surface expression of virulence factors by nonmucoid BCC strains [63]. The switch to mucoidy in BCC has been attributed to a more metabolically dormant and less aggressive phenotype. Overall, however, BCC infection confers a poor prognosis [64-67], and BCC has been suggested to outcompete P. aeruginosa in CF lungs. This advantage may be due to a primary siderophore, ornibactin, that is far more effective at obtaining iron from the host than the two primary P. aeruginosa siderophores, pyoverdine and pyochelin [68].
Other pathogens, including H. influenzae and S. maltophilia, are also frequent CF lung colonizers, with prevalence rates of 15.5 and 13.6%, respectively. H. influenzae, a gram-negative coccobacillus, is sometimes the earliest infectious organism recovered from very young CF patients, and causes chronic inflammation similar to P. aeruginosa [69,70]. It has been suggested that infection by H. influenzae (and consequent inflammation) early in life might increase susceptibility to infection by P. aeruginosa [71]. The prevalence of S. maltophila, a gram-negative bacillus, appears to have increased due to use of anti-pseudomonal drugs [72]. These bacteria are emblematic of a pathogenically significant microbiome that includes many organisms of unknown pathogenic significance. As certain niches are emptied over the course of a CF patient's lifetime, new bacteria adapt to inhabit these microenvironments.
Similar to CF, the lungs of individuals with COPD are chronically infected yet with many similarities and differences. Although S. pneumoniae, Haemophilus influenzae and Moraxella catarrhalis are the predominant pathogens, others that have been identified include Mycoplasma pneumoniae, P. aeruginosa, Citrobacter freundii, S. aureus, Enterobacter cloacae, Stenotrophomonas maltophilia, Klebsiella pneumoniae, Proteus mirabilis, and Serratia marcescens [73]. Viruses identified include parainfluenza virus, influenza virus, RSV, and rhinovirus.
M. catarrhalis and S. pneumoniae are two pf the most frequent species commonly cultured from the lungs of individuals with COPD, although their prevalence is less than in CF. A gallery of similar pathogens is found in COPD compared with CF, although the dominant organisms are different. Non-typable H. influenzae (NTHi) is the most common infectious bacterium observed in COPD and colonizes 60% of COPD patients [23,74-79]. During acute exacerbations, NTHi is the most likely bacterium to be found in the airway [74-78]. Since NTHi is cultured from individuals with stable COPD as well as during exacerbations, it has been suggested that these bacteria stimulate an inflammatory response in both clinical scenarios, and exacerbations tend to be more severe when NTHi is present. In addition, acquisition of new strains of NTHi increase the risk of frequent exacerbations [80-82]. NTHi has the ability to avoid clearance from the lungs contributing to its status as a refractory pathogen. The organism uses outer membrane proteins P2 and P5 to facilitate bacterial binding to respiratory mucus by lipooligosaccharide (LOS), a low molecular weight version of the more typical bacterial LPS. This pathogenic mechanism causes ciliary dysfunction, diminishing mucus clearance [83,84]. To further defend itself during infection, NTHi also secretes IgA proteases that bind and degrade IgA (the major antibody in mucosal secretions), reducing levels of IgA in the airway lumen, and, thereby, decreasing the ability to clear the organism. This adaptation not only allows NTHi to flourish, but also promotes growth and airway colonization of other pathogens, leading to complex infections that are difficult to eradicate. The same interactions have been reported after NTHi infection in CF lungs [85,86].
It is interesting to note that M. catarrhalis, a gram-negative diplococcus and commensal organism in the upper respiratory tract of humans was not initially deemed a pathogen in COPD. This bacterium was isolated frequently from the sputum of COPD patients, but its pathogenic capacity was not recognized until the early 1990's [87-89]. Since then, the organism has been established as a major cause of lung infections in COPD and a leading cause of exacerbations [90-93]. By adhering to epithelial cell surfaces, M. catarrhalis is able to persist in the lungs and elicit chronic infection. This propensity is stimulated by host immune defensins [94]. With a robust immune response, M. catarrhalis is stimulated to adhere to the cell surface, mediated by UspA, which binds to carcinoembryonic antigen-related cell adhesion molecules at the epithelial cell plasma membrane [95]. This interaction further promotes an airway inflammatory response. Along with the ability to adhere, ~90 percent of M. catarrhalis strains found in the lower respiratory tract resist complement-mediated killing by the immune system by virtue of a disulfide bond formation system that helps stabilize the lipopolysaccharide resisting complement attack [96,97]. Despite this survival mechanism, the organism remains susceptible to most antibiotics used to treat respiratory tract infection. An exception is resistance to trimethoprim and ß-lactams, which occurs through naturally insensitive dihydrofolate reductase enzymes and the production of a ß-lactamase [98-101].
Another common pathogen in COPD lungs is the gram-positive coccus, S. pneumoniae, typically found in the respiratory tract during both periods of both stability and exacerbation. As many COPD exacerbations are associated with bacterial lung infections, patients with sputum cultures revealing S. pneumoniae are not infrequently placed empirically on antibiotics stimulating a higher prevalence of antibiotic resistance among pneumococcal species [102]. S. pneumoniae is known to cause both exacerbations and an increased risk for pneumonia in patients with COPD [103,104]. Acute exacerbation is elicited by bacterial virulence factors and the immune response to new infection. An important virulence factor in this setting is the polysaccharide capsule that mediates evasion from immune clearance. Capsular features may be helpful in identifying pathogenic potential of various pneumococcal species [105].
The presence of S. pneumoniae confers a higher risk of exacerbation in COPD, but interestingly only when cultured in the absence of other pathogens. In mixed culture, the risk of exacerbation does not appear to be elevated, which suggests that singular culture represents a more virulent species [106]. Pneumococcal vaccines help reduce invasive infections caused by the many prevalent S. pneumoniae serotypes by inducing an adaptive immune response [107,108].
Although P. aeruginosa causes chronic respiratory infection in COPD, it occurs much less frequently than in CF. Still the organism is associated with considerable levels of respiratory impairment [109-112] and is isolated in 6% of acute COPD exacerbations. An increased prevalence of multi-drug resistant strains is observed in critically-ill patients [110,111,113]. Exacerbations can be most readily attributed to P. aeruginosa during acquisition of a new strain which elicits an exuberant immune response [114]. The immune system responds by stimulating additional virulence factors from the P. aeruginosa as well as further inflammation. Moreover, the presence of the mucoid phenotype may be observed in this setting, and, just as in CF, these mucoid strains persist in the lungs as biofilms (Figure 8), while non-mucoid strains may not. The mucoid P. aeruginosa phenotype less common in COPD compared with CF, and the estimated prevalence's are 8% and 48%, respectively [115].
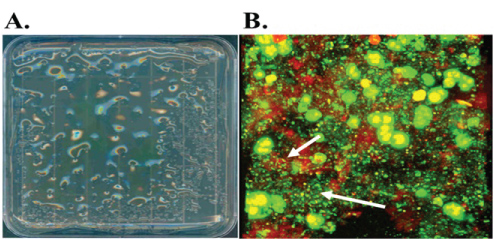 Figure 8: A. L-agar plate of mucoid P. aeruginosa derived from a chronically infected COPD patient. B. Confocal laser scanning micrograph of sputum from the same patient with live (green)/dead (red) staining.
View Figure 8
Figure 8: A. L-agar plate of mucoid P. aeruginosa derived from a chronically infected COPD patient. B. Confocal laser scanning micrograph of sputum from the same patient with live (green)/dead (red) staining.
View Figure 8
The biochemical and cellular derangements of CF produce innate immune system dysfunction. A normal component of innate lung defense is mechanical clearance of airway secretions by cilia on the epithelial cell surface [116-118]. Viscous mucus has several major consequences in the airway. First, as noted above, mucus compresses cilia against the cellular surface, and inhibits proper ciliary activity. Second, due to an already decreased clearance capacity, mucus directly interacts with the epithelial cell membrane. Over time, concentrated mucins directly anneal to the epithelial layer, and cannot be cleared by the cilia or by natural mechanical disruption (e.g. coughing or chest physical therapy) [119]. These factors contribute to the characteristic mucus stasis and inflammation in the CF lungs A build-up of impacted mucus often begins at birth and continues throughout life in individuals with CF [120,121]. If it is not cleared, mucus forms an ideal niche that permits colonization by opportunistic microorganisms. Mucus plaque formation provides a surface on which bacteria adhere and form biofilms, which further increases plaque surface exopolysaccharide content, establishing a cyclical process of bacterial adherence, biofilm formation, and mucus plaque accumulation (Figure 9).
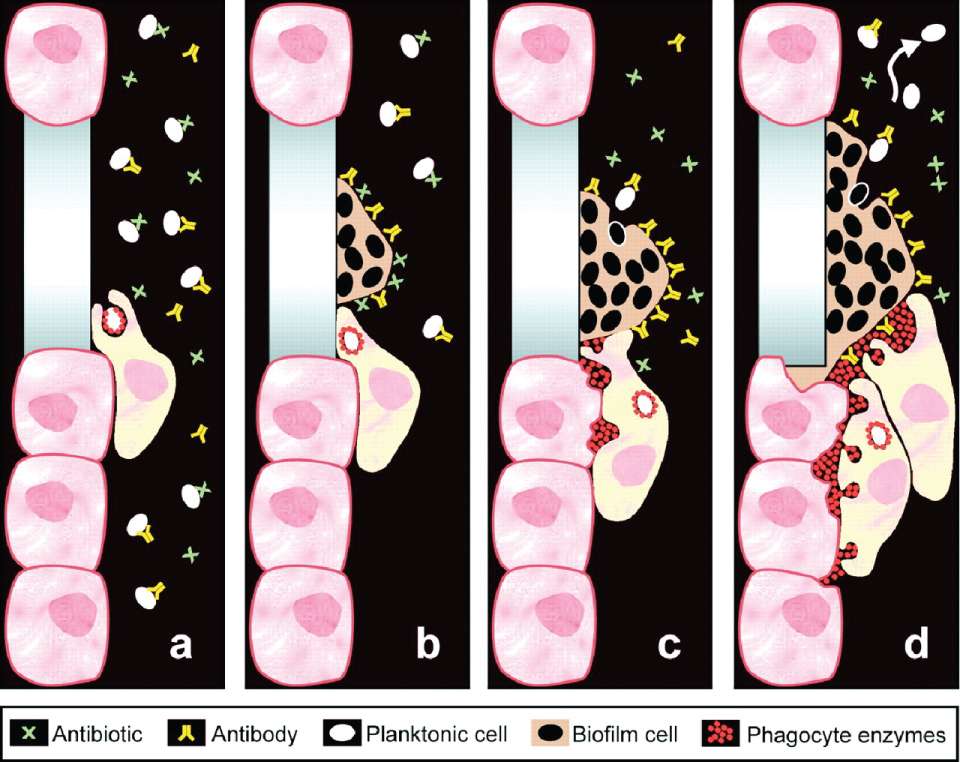 Figure 9: Biofilms promote bacterial persistence during treatment. Planktonic bacteria can be cleared by antibiotics, antibodies, or host human cells. Once a biofilm has formed, these elements may become less effective. Enzymes utilized as part of phagocytosis build up within host cells and elicit cell damage and increased inflammation. If bacteria return to planktonic form, the immune system and antibiotics are able to more effectively address infection. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070663094.
View Figure 9
Figure 9: Biofilms promote bacterial persistence during treatment. Planktonic bacteria can be cleared by antibiotics, antibodies, or host human cells. Once a biofilm has formed, these elements may become less effective. Enzymes utilized as part of phagocytosis build up within host cells and elicit cell damage and increased inflammation. If bacteria return to planktonic form, the immune system and antibiotics are able to more effectively address infection. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070663094.
View Figure 9
A second underlying problem with immune responsiveness in CF patients is the abnormal degradation and cell trafficking of Toll-like Receptor 4 (TLR4). Normally, TLR4 receptor is present within the Golgi [122,123] and may migrate to the cell surface and bind LPS as part of a receptor complex that assembles on lipid rafts and activates NF-KB and MAPK pathways [124,125]. Subsequently, the receptor is ubiquitinated and becomes associated with endosomes, where it activates INFR3 [122,126,127]. These events contribute to the degradation of TLR4, and even subtle changes in this mechanism can perturb the immune response [122,128-130]. Abnormal trafficking of TLR4 leads to increased LPS-induced activation involving multiple components of immune activity, including NF-KB, MAPK signaling, and IFN regulatory factor-3 (IFNR3) [131]. Along with increased immunological responsiveness, these events may decrease TLR4 degradation, which further disrupts airway defense. Macrophages from CF patients may be hyper-responsive to bacterial LPS [132], due, in part, to abnormal TLR4 trafficking [132].
CF also dramatically affects function and accumulation of phagocytes in lung tissues [133]. Neutrophils, which accumulate to nearly 1500-fold above their normal levels, have impaired migration through the mucus in an attempt to clear bacteria before refractory biofilms are established, which may allow changes in the bacterial phenotype, including mucoid conversion of P. aeruginosa [133-135]. Ineffective attempts at bacterial killing by neutrophils increase DNA deposition associated with neutrophil extracellular traps (NETs), which further contribute to mucus viscosity. Not only is the ability of neutrophils to phagocytose bacteria compromised, but steep oxygen gradients established by pathogens in airway secretions significantly impact generation of microbicidal reactive oxygen species (ROS) [135]. Without ROS (or reactive nitrogen species), neutrophil function is substantially compromised.
Unlike CF airway, disease cigarette smoking is the major etiologic factor contributing to the development of COPD and this exposure elicits multiple innate immune system derangements (Figure 10). Cigarette smoke (CS) directly impairs mucociliary clearance [136], including both ciliary shortening and physiologic function [137-139]. Direct cell death from CS exposure also leads to re-epithelialization that is dominated by goblet cells, a cellular compartment associated with mucus production [140,141]. Shortened cilia after CS exposure are associated with histone deacetylase 6-mediated selective auto phagocytosis and further degradation of cilia [136]. Chronic reduction in mucociliary clearance promotes susceptibility to bacterial infection in patients with COPD, just as in CF.
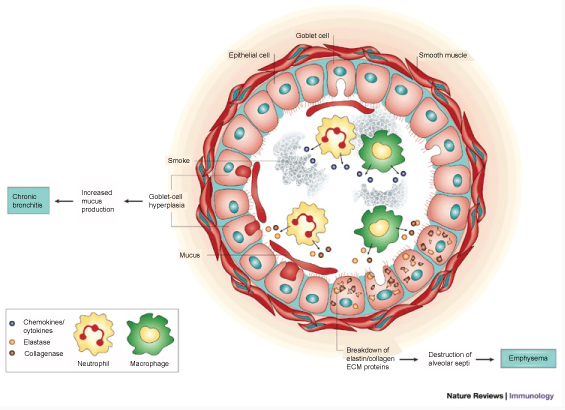 Figure 10: Smoking and the Immune System. With permission from [194]. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070475915.
View Figure 10
Figure 10: Smoking and the Immune System. With permission from [194]. Permissions from Copyright Clearance Center - Rightslink, order number: 4336070475915.
View Figure 10
CS also affects resident immune cells of the lung. These effects include increased numbers of alveolar macrophages and reduced ability to clear apoptotic cells and bacterial infections, due to impaired monocyte differentiation and lowered expression of surface recognition molecules [142,143]. CS also increases expression of pro-inflammatory chemokines and matrix metalloproteases, which suggest a change in macrophage chemokine phenotype [144,145]. Neutrophil ROS production regulates the phagocytic respiratory burst, and phagocytosis impairment during differentiation is another factor contributing to reduced bacterial clearance in COPD [146]. Failure of neutrophil function is compounded by reduced antimicrobial capacity of macrophage apoptosis. The polymorphonuclear cell derangements in COPD may also increase extravasation of lysozymes and granules into the extracellular space, contributing to pulmonary structural damage [147].
Activity of natural killer (NK) cells, which normally contribute to eradication of viral pathogens, is increased by CS. Elevated expression of epithelial cell surface ligands is associated with CS exposure and stimulates the NK cell by binding to the NKG2D receptor. These activated NK cells may promote airway epithelial cell apoptosis and tissue damage due to dysregulated inflammation [148].
Key aspects of CF treatment have traditionally focused on addressing symptoms of the disease, but more recently have included interventions directed towards correcting fundamental physiologic abnormalities caused by mutation of CFTR. Symptomatic or palliative treatments, for example, include compensation for pancreatic insufficiency with supplemental pancreatic enzymes, high calorie diets with inclusion of fat-soluble vitamins, and anti-inflammatory agents to slow progression of respiratory function decline. General interventions for lung disease also encompass chest physical therapy and inhaled treatments to improve mucus clearance, together with antibiotic therapy for infection control [149].
Failure of mucus clearance is a hallmark of CF pathogenesis, and a number of treatments have been developed to overcome this defect. Mechanical devices and patient compliant actions (chest physical therapy, aerobic exercise, etc.) increase mucus mobilization, and are part of standard CF clinical care. Treatments include use of active cycle breathing techniques and autogenic drainage, a breathing technique used to mobilize mucus up the airway, where it can be more easily cleared by coughing. Positive expiratory pressure masks and high frequency chest wall oscillation can aid in this process [150,151]. Prescription of these methods is typically provided on an individualized basis, as there is no evidence that one technique works more effectively in all cases [152]. Furthermore, it is not established that use of airway clearing techniques is beneficial in the early stages of CF, when there may be little sign of lung impairment, and build-up of mucus is less pronounced. That being said, recent treatment guidelines often recommend daily airway clearance and aerobic exercise to help improve mucus clearance as a means to improve patient health [153].
Mucolytic compounds are used to breakdown excess mucus lining the airways. In addition to airway secretions, themselves, DNA from neutrophil extracellular traps contributes significantly to increased CF sputum viscosity. Use of recombinant human DNase, such as dornase alfa, can be used to decrease viscosity and augment lung function [154,155]. Another useful mechanism is increased hydration of airway secretions. Inhaled agents such as hypertonic saline (7%) stimulate movement of vascular water into thick airway secretions, helping cilia mobilize sputum and promote cough-mediated clearance [156-158].
Newer modes of treatment aim to target basic genetic defects responsible for CF [159]. One such technique is to bypass or repair DNA and/or mRNA encoding mutant CFTR protein. Approaches using viral vectors - e.g. adeno-, adeno-associated, or retro-viruses - were used to insert functioning copies of the CFTR gene into airway epithelia. While early attempts towards CFTR replacement led to inadequate gene transmission and immune responses upon repeated administration [160], technology in this area has continued to advance. Repeated nebulization of plasmid DNA and liposome complex [161] in a double-blind study showed modest stabilization of lung function when the test group was compared to the control after one year. Adverse events were noted in both study cohorts, with more serious effects observed after plasmid treatment. Gene transfer approaches such as these, as well as newer viral delivery vehicles, represent important areas for future investigation.
More successful methods that aim to treat the underlying genetic cause of cystic fibrosis act on the mutant protein directly. One example is the combination of lumacaftor and ivacaftor, two molecules that target the classic D508 variant [159]. Lumacaftor is an agent known as a 'corrector'; it has been shown to partially 'correct' misprocessing of the F508del mutant, increasing its presence at the cell surface [162]. This alone does not lead to significant effects on disease severity, but in combination with ivacaftor, an FDA approved 'potentiator' (activator of ion channel gating), significant clinical benefit has been demonstrated among F508del/F508del homozygous individuals. Ivacaftor acts to increase the probability that the CFTR channel is open, allowing for chloride and bicarbonate movement and proper function [163]. The drug combination (ivacaftor together with lumacaftor) led to improvement of FEV1 in patients homozygous for the F508del mutation, representing a significant breakthrough (applicable to ~40% of individuals with CF [164]. Ivacaftor as a single drug has also shown robust benefit among numerous partial function CFTR mutations, for which the compound is FDA approved.
Strategic antibiotic regimens are commonly used to control infection of CF airways, although resistance has become an increasing issue. A large subset of these target P. aeruginosa. Nebulized antibiotics including tobramycin, colistin and others are routinely administered and reach high concentrations in lower trachea and upper airways; penetration to the most distal airways may be insufficient [165-169]. In comparison, when antibiotics are given intravenously or orally, drugs are delivered to the deep respiratory tract via the pulmonary circulation, but may be inadequately transferred to sputum, due partly to CFTR-mediated secretory and mucus viscosity issues [170]. The combination of both routes is essential since P. aeruginosa is established throughout the lung. [29,171,172]. Development of resistance is common; P. aeruginosa (and other bacteria such as BCC) adapt through formation of biofilms, compounding issues related to chronic infection [173].
Dispersal of thick bacterial biofilms is an important step towards treating CF lung infection, since antibiotics are far more efficacious (by 10-100-fold) against planktonic (free-living) P. aeruginosa. Biofilms that arise after the ∆mucA mutation are inherently resistant to antibiotics and phagocytic neutrophils [174,175]. One experimental treatment that has been tested in vitro and in vivo (mouse chronic infection model) uses acidified nitrite (NO2-) administration at pH 6.5 [176]. The acidic pH reflects that of CF lungs, and bacterial killing is pH dependent. Formation of HNO2 and NO through this approach may enhance NO associated with anaerobic respiration of the organism in adherent CF mucus. Antibiotic resistant strains were found to be highly sensitive to HNO2, indicating importance of further studies in this area.
A vitally important treatment for CF involves lung transplantation. Infants and toddlers with CF comprise a minority of healthy lung recipients [177]. Liou, et al. used retrospective data to show that pediatric patients may benefit less from the intervention [178], whereas other studies have disputed these findings [179].
Since nearly all the lung damage that occurs in COPD cannot be reversed once it occurs, the primary goal is disease prevention. Treatment strives to minimize respiratory symptoms and complications, maintain lung function, and preserve quality of life. Management can be achieved through pharmacological or non-pharmacological means.
Multiple drug classes are utilized as interventions for COPD. Short acting beta agonists (SABA) represent the initial and most frequently used medications applied for wheezing or breathlessness. These drugs bind to the β-adrenergic receptor, stimulating smooth muscle cell relaxation and airway dilation. Long acting anticholinergics comprise a mainstay of chronic maintenance treatment for COPD, and mitigate many COPD symptoms, including airflow limitation and dynamic hyperinflation, acute COPD exacerbations, and lung function deterioration [180-182]. Long acting beta agonists may be used alone or in combination with long acting anticholinergics. A third class of medication is inhaled corticosteroids (ICS). ICS are recommended when FEV1 is lower than 50% among patients who have had two or more exacerbations in the prior year. ICS may be administered in combination with long acting beta agonists and long acting anticholinergics. This triad of drugs may improve lung function but does not reduce exacerbations when compared to other modalities [183]. Approximately 70% of COPD patients receive ICS, but only 10% may actually qualify according to current guidelines [184]. Phosphodiesterase (PDE) inhibitors block breakdown of signaling molecules (such as cAMP and cGMP). This process reduces inflammation and stimulates bronchodilation. Roflumilast reduces the number of exacerbations in patients with severe COPD-associated bronchitis and recurring exacerbations [185].
As with CF, pharmacological treatments utilized in COPD also include mucolytics and antibiotics. Mucolytics cleave respiratory secretions and have been reported to improve overall quality of life [186]. Cleaved mucus is more readily mobilized from lungs with damaged cilia. Improved clearance reduces ability of bacteria to bind to the airway epithelial surface and promotes neutrophil activity, which helps quell the exuberant immune response in COPD. Despite its beneficial effects in CF, rhDNase is detrimental in individuals with COPD and reduces lung function and increases exacerbations [187]. Other mucoactive agents such as normal mannitol, saline, and hypertonic saline may cause transient bronchospasm, cough, and dyspnea upon initiation but may have slight beneficial clinical and physiologic effects [187]. Because exacerbations are frequently triggered by bacterial infection, antibiotics are often used in a manner similar to that described for CF. Chronic macrolide use reduces the likelihood of COPD exacerbation, an effect believed to be mediated by macrolide anti-inflammatory properties, rather than its antibacterial effects. Importantly, prolonged use of either erythromycin or azithromycin may lead to the selection of resistant organisms [188].
A critical, non-pharmacological treatment modality for COPD involves smoking cessation. Nicotine replacement is commonly used; other smoking cessation medications include bupropion and partial nicotinic receptor agonists such as varenicline. Promising reductions in smoking have been achieved with varenicline. Finally, treatments that prolong life in individuals with COPD include supplemental oxygen in the setting of resting hypoxemia, vaccinations, pulmonary rehabilitation, and lung volume reduction procedures [189].
Chronic respiratory diseases including COPD and CF are the third leading cause of death in the United States currently. Both disorders provoke similar respiratory symptoms and can lead to respiratory insufficiency and death. Common morbidities include bacterial infections caused by similar bacteria; common pathophysiologic processes include mucostasis and abnormalities of the innate immune system. Further research is needed to achieve the goal of managing these disorders and ultimately prolonging lives of both patient populations.
This work was funded in part from the Department of Veteran’s Affairs, Cincinnati VA Medical Center Merit Award, #5I01BX000845-03 (D.J.H.), National Institutes of Health (R01HL136414, R01HL139876 [E.J.S.], R01HL116226 [J.P.C.], Cystic Fibrosis Foundation (CFF SORSCH13XX0, CFF SORSCH14XX0) [E.J.S.].