

International Journal of Surgery Research and Practice
A Rare Case of Biliary Atresia and Choledochal Cyst in a Premature Infant
Abigail B Podany1, Ryan M Juza1, Sosamma T Methratta2, Peter W Dillon3, Mary C Santos3, Brett W Engbrecht3, Robert E Cilley3, Dorothy V Rocourt3 and Karmaine A Millington4
1Department of Surgery, Penn State Milton S. Hershey Medical Center, USA
2Department of Pediatric Radiology, Penn State Milton S. Hershey Children's Hospital, USA
3Department of Surgery, Penn State Milton S. Hershey Children's Hospital, USA
4Department of Pathology, Penn State Milton S. Hershey Medical Center, USA
*Corresponding author: Dorothy V Rocourt, MD, Assistant Professor of Surgery and Pediatrics, Department of Pediatric Surgery, Penn State Hershey Children's Hospital, 500 University Drive, H1113, Hershey, PA 17033, USA, Tel: (717) 531-8342, Fax: (717) 531-4185, E-mail: drocourt@hmc.psu.edu
Int J Surg Res Pract, IJSRP-3-037, (Volume 3, Issue 1), Case Report; ISSN: 2378-3397
Received: November 14, 2015 | Accepted: February 26, 2016 | Published: February 28, 2016
Citation: Podany AB, Juza RM, Methratta ST, Dillon PW, Santos MC, et al. (2016) A Rare Case of Biliary Atresia and Choledochal Cyst in a Premature Infant. Int J Surg Res Pract 3:037. 10.23937/2378-3397/1410037
Copyright: © 2016 Podany AB, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Recent case reports have described variants in the classification of biliary atresia and choledochal cyst requiring the expansion of the classical system for describing these anomalies. This in effect, further obscures the line between these two previously separate entities while highlighting the difficulties of intraoperative differentiation of biliary atresia, cystic biliary atresia, choledochal cyst, and Caroli's disease. In our case report, we describe a 34-week premature infant who developed a direct hyperbilirubinemia and was found to have an atypical presentation of choledochal cyst and biliary atresia. We believe this case demonstrates the difficulty in distinguishing and managing this disorder.
Keywords
Biliary atresia, Choledochal cyst, Caroli's disease
Case Report
Our patient was a 34-week, 3.2 kilogram premature but otherwise healthy-appearing African American male infant with no significant family history. After a short hospital stay during which the patient was noted to be hyperbilirubinemic, he was discharged home on day of life (DOL) five. While at home the patient remained jaundiced and developed acholic stools. He was evaluated by Pediatric Gastroenterology and worked up with hepatobiliary iminodiacetic acid (HIDA) scan and ultrasound (Figure 1), which were consistent with choledochal cystand possible biliary atresia. The patient was referred to Pediatric Surgery for definitive management.

.
Figure 1: Preoperative Ultrasound (A) and HIDA scan (B) demonstrating a cyst at the portahepatis, absent gall bladder, and biliary outflow obstruction.
View Figure 1
Operative Approach
The patient was taken to the operating room on DOL 28. At exploration, the gallbladder was noted to be atretic with clear mucus. The liver was somewhat firm in texture and dark red-grey in color. There was no obvious cirrhosis or fibrosis. Liver biopsies were taken and sent for pathology. At the confluence of the cystic and hepatic ducts, a 2-3 cm cyst was noted with obliteration of the distal common bile duct as it extended posterior to the duodenum. Choloangiogram first performed through the gallbladder and then through the cyst did not demonstrate proximal filling of ductal structures. The cyst was mobilized and was sharply transected just below the bifurcation of the right and left hepatic ducts. Copious bile flow was noted from the proximal hepatic duct stump. The proximal hepatic stump appeared to be of normal ductal size measuring approximately 2-3 mm in transverse diameter. This tract was easily intubated and cholangiogram demonstrated abnormal, but patent right and left hepatic ductal system with radicals (Figure 2). Because the diagnosis of biliary atresia versus choledochal cyst had not been entirely eliminated, the decision was made to reconstruct the patient's biliary outflow with a hepaticoduodenostomy in the event that it may be necessary to re-operate to perform a hepatoportoenterostomy [1]. A short segment of 3.5 mm feeding tube was used as a stent in the hepatic duct and a hepaticoduodenostomy created.
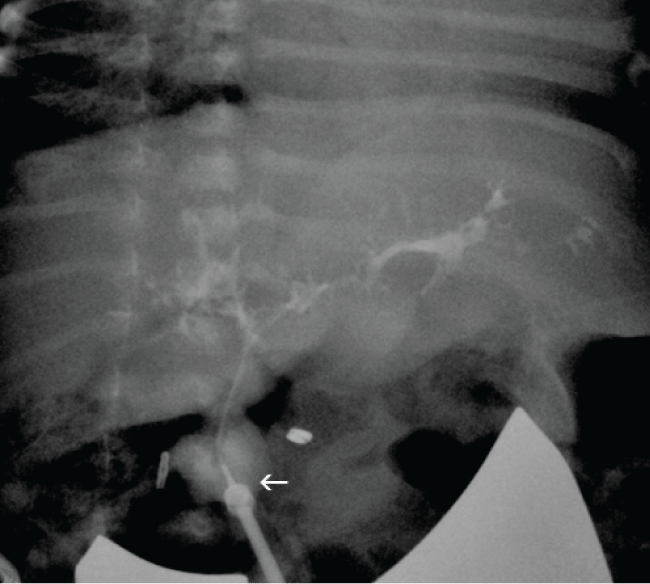
.
Figure 2: Intraoperative cholangiogram through proximal hepatic duct demonstrating ectatic but patent intrahepatic ducts. Arrow indicates catheter in hepatic duct.
View Figure 2
On histologic examination of the liver wedge biopsies taken at the initial operation, fibrosis in mainly a portal distribution was evident, and bile ductular proliferation could be seen in most of the portal tracts. The excess bile that accumulated secondary to downstream obstruction formed bile plugs in canaliculi and in the ductules. The resected cyst measured 1.5 × 1.2 × 0.8 cm with a wall thickness of 0.6 cm and a lumen devoid of contents (Figure 3A).
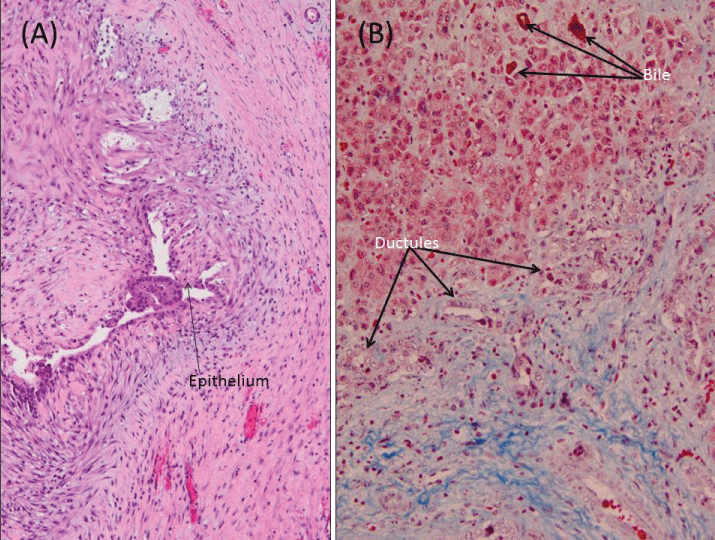
.
Figure 3: Liver Histopathology. (A) Hematoxylin& Eosin, 100X magnification- wall of choledochal cyst, with epithelial denudation, inflammation, and reactive hyalinization; (B) Trichrome stain, 200X magnification- liver biopsy from Kasai hepatoportoenterostomy with marked fibrosis, proliferation of bile ducts, and cholestasis.
View Figure 3
Postoperatively the patient did well. The patient had return of bowel function and tolerated advancing his diet. His intra-abdominal drain was discontinued. Total bilirubin however, remained elevated and steroids were started one week postoperatively in an effort to limit progressive fibrosis of the biliary tree [2,3]. His hyperbilirubinemia persisted. Given the persistent hyperbilirubinemia and pathologic findings consistent with biliary atresia, the decision was made to take the patient back to the operating room for a formal Kasai portoenterostomy.
Second Operative Course
The patient was brought back to the operating room on DOL 70. His original incision was reentered and the hepaticoduodenostomy was taken down. The duodenotomy was closed transversely. Dissection was then carried down toward the porta hepatis. A rather broad liver plate was identified and sharply incised at which point good bile flow was noted. A hepatoportoenterostomy was then created using a mobilized antecolic jejunal conduit. The liver biopsy performed at the time of the Kasai portoenterostomy demonstrated progression of the fibrosis, and the common right and left hepatic ducts resected were markedly narrowed, the central lumen obscured by fibrosis. Denudation of the epithelium proximally was followed in the distal region by focal absence of the lumen (Figure 3B).
The patient's second postoperative course was protracted. While his bilirubin trended down gradually, he had slow return of bowel function and persistent abdominal distention. A HIDA scan demonstrated biliary drainage without evidence of anastomotic leak.
Several weeks after his second operation he was readmitted with fevers and feeding intolerance. On workup he was found to have new cavitary lesion consistent with abscess of the left hepatic lobe (Figure 4A). A percutaneous drain (Angiotech, Vancouver, BC) was placed and a study through the drain demonstrated continuity with the left hepatic duct (Figure 4B). This was therefore felt to be infected biliary abscess rather than bile lake formation. The patient subsequently developed an anastomotic stricture which was amendable to drainage catheter placement through the percutaneous drain (Cook Medical, Bloomington, IN). This drain was later internalized and upsized from 5-French to 6.5-French to maintain the patency of his anastomosis. After several weeks, the catheter was removed and drain study demonstrated patent intrahepatic ducts. All drains were therefore removed. At the recommendation of gastroenterology, he continues to be on prophylactic Ursodeoxycholic acid and sulfamethoxazole/trimethoprim. He is now four years out from his last surgery. He is followed biannually by gastroenterology, and annually by pediatric surgery. He has had no episodes of jaundice, cholangitis, or changes in stooling pattern. His bilirubin levels have remained normal. He is at the 95th percentile for weight.
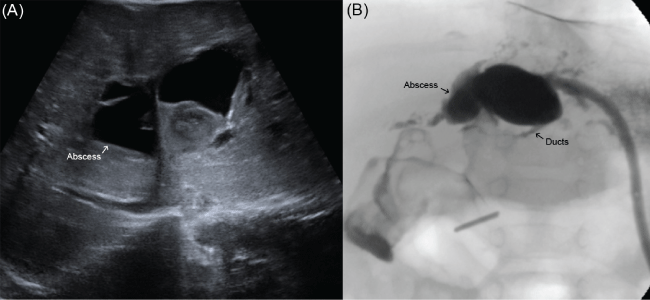
.
Figure 4: (A) Ultrasound demonstrating abscess in the left hepatic lobe; (B) Drain study through percutaneous drain demonstrating continuity between the cavity and the main left hepatic duct.
View Figure 4
Discussion
Surgical correction is required in the treatment for pediatric biliary atresia and choledochal cyst. Age at initial operation has been shown to have the most significant effect on overall survival and subsequent need for liver transplant [4-6]. Recently, there have been multiple articles published, highlighting the difficulty in distinguishing between variants of biliary atresia and choledochal cyst in pediatric hepatobiliary surgery [4,5,7,8]. The adjunct of specific ultrasound findings have been described to help differentiate biliary atresia from choledochal cyst are supportive but not definitive [9]. Regardless, successful surgical management of this disorder requires accurate initial differentiation between choledochal cyst and biliary atresia. Attempts to establish radiographic diagnostic criteria have not been highly successfully, and the mainstay of diagnosis still includes surgical exploration for pathology and cholangiography [4,10].
In our case report, the patient was diagnosed early in life with obstructive jaundice. However, as has previously been reported in other patients, imaging studies were inconclusive in differentiating between choledochal cyst and biliary atresia [10,11].Despite this, our patient underwent operative management in a timely fashion. Liver parenchymal biopsies taken at the beginning of the operation were consistent with biliary atresia. Intraoperatively, a cystic structure was identified at the confluence of the cystic and hepatic ducts giving the appearance of a Type I choledochal cyst with distal atresia. This diagnosis was further reinforced whereupon transecting the proximal hepatic duct, good bile flow was noted and cholangiogram was able to demonstrate patent right and left hepatic ducts with radicals. Our patient failed reconstruction with hepaticoduodenostomy and eventually required formal portoenterostomy as he had progression of fibrosis. Kasai portoenterostomy remains first-choice procedure. In the long term, patients with biliary atresia may go on to require liver transplantation, even into adulthood [12].
Conclusions
Despite good evidence to support the diagnosis of Type I choledochal cyst with distal atresia, our patient failed reconstruction with hepatoduodenostomy and eventually required a second operation with formal Kasai portoenterostomy [13]. We believe that this case highlights the difficulties in making the diagnosis in the setting of congenital anomalies of the pediatric biliary tree and underscores the importance of surgical exploration and pathologic diagnosis. It is unclear whether the patient's phenotype was a variant in the spectrum of biliary atresia and choledochal cyst or whether this infant's prematurity created an evolution of postnatal progressive fibrosis, which ultimately led to failure of his initial reconstruction. Recent case reports have described the caveats of diagnosing biliary atresia in the setting of choledochal cyst, which correlate with our report [11,14,15]. Despite postoperative setbacks following his second operation, the patient is doing well and liver functions have stabilized near normal at four years of age.
Acknowledgements
The authors would like to acknowledge Dr. Allene Burdette and the department of Pediatric Radiology at Penn State Children's Hospital for assistance performing and interpreting radiologic studies.
Disclosures
The authors have nothing to disclose.
References
-
Narayanan SK, Chen Y, Narasimhan KL, Cohen RC (2013) Hepaticoduodenostomy versus hepaticojejunostomy after resection of choledochal cyst: a systematic review and meta-analysis. J Pediatr Surg 48: 2336-2342.
-
Dillon PW, Owings E, Cilley R, Field D, Curnow A, et al. (2001) Immunosuppression as adjuvant therapy for biliary atresia. J Pediatr Surg 36: 80-85.
-
Zhang D, Yang HY, Jia J, Zhao G, Yue M, et al. (2014) Postoperative steroids after Kasai portoenterostomy for biliary atresia: a meta-analysis. Int J Surg 12: 1203-1209.
-
Caponcelli E, Knisely AS, Davenport M (2008) Cystic biliary atresia: an etiologic and prognostic subgroup. J Pediatr Surg 43: 1619-1624.
-
Davenport M (2012) Biliary atresia: clinical aspects. Semin Pediatr Surg 21: 175-184.
-
Wildhaber BE, Coran AG, Drongowski RA, Hirschl RB, Geiger JD, et al. (2003) The Kasai portoenterostomy for biliary atresia: A review of a 27-year experience with 81 patients. J Pediatr Surg 38: 1480-1485.
-
De Matos V, Erlichman J, Russo PA, Haber BA (2005) Does "cystic" biliary atresia represent a distinct clinical and etiological subgroup? A series of three cases. Pediatr Dev Pathol 8: 725-731.
-
Arora A, Patidar Y, Khanna R, Alam S, Rastogi A, et al. (2012) Cystic biliary atresia: confounding and intriguing. J Pediatr 161: 562.
-
Zhou LY, Guan BY, Li L, Xu ZF, Dai CP, et al. (2012) Objective differential characteristics of cystic biliary atresia and choledochal cysts in neonates and young infants: sonographic findings. J Ultrasound Med. 31: 833-41
-
Jiexiong F, Minju L, Hongfeng T, Weizhong G, Shaoyong Y (2003) Clinical and pathological characteristics of cystic lesions of extrahepatic bile duct in neonates. Acta Paediatr 92: 1183-1189.
-
Sinha S, Sarin YK (2013) Extra hepatic biliary atresia associated with choledochal cyst: a diagnostic dilemma in neonatal obstructive jaundice. J Neonatal Surg 2: 11.
-
Segura-Sampedro JJ, Bernal-Bellido C, Marín-Gómez LM, Suárez-Artacho G, Serrano-Díez-Canedo J, et al. (2015) Outcomes of Liver Transplantation During Adulthood After Kasai Portoenterostomy Due to Biliary Atresia. Transplant Proc 47: 2643-4.
-
Mendoza MM, Chiang JH, Lee SY, Kao CY, Chuang JH, et al. (2012) Reappraise the effect of redo-Kasai for recurrent jaundice following Kasai operation for biliary atresia in the era of liver transplantation. Pediatr Surg Int 28: 861-864.
-
Mourier O, Franchi-Abella S, Ackermann O, Branchereau S, Gonzales E, et al. (2011) Delayed postnatal presentation of biliary atresia in 2 premature neonates. J Pediatr Gastroenterol Nutr 52: 489-491.
-
Sookpotarom P, Vejchapipat P (2007) Non-correctable biliary atresia with large extrahepatic cyst: a report of two cases. Eur J Pediatr Surg 17: 295-297.
