

Journal of Genetics and Genome Research
Data-driven Biomarker and Drug Discovery using Network-based Approach
Fuhai Li* and Ming Zhan
Department of Systems Medicine and Bioengineering, Houston Methodist Research Institute, Weill Cornell Medical College, Houston, Texas, USA
*Corresponding author:
Fuhai Li, Department of Systems Medicine and Bioengineering, Houston Methodist Research Institute, Weill Cornell Medical College, Houston, Texas, USA, E-mail: fli@houstonmethodist.org; robert.fh.li@gmail.com
J Genet Genome Res, JGGR-2-020, (Volume 2, Issue 2), Review Article; ISSN: 2378-3648
Received: May 11, 2015 | Accepted: November 02, 2015 | Published: November 05, 2015
Citation: Li F, Zhan M (2015) Data-driven Biomarker and Drug Discovery using Network based Approach. J Genet Genome Res 2:020. 10.23937/2378-3648/1410020
Copyright: © 2015 Li F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
An increasing body of large-scale genomic profiling data has been being generated on many diseases including cancers and on a number of drugs and compounds. The exploration of such big data has led to data-driven biomedical research. The data-driven studies include exploring disease subtypes with distinct molecular patterns, uncovering novel diagnosis biomarkers or treatments, and discovering new indications of drugs along with novel mechanisms of drug action, among others. However, challenges remain to integrate, interpret and convert the big biomedical data into informative knowledge or therapeutic discovery, and demanding sophisticated computational approaches for the big data analysis. Here, we review the network-based approaches as a promising strategy for novel biomarker and drug discovery of cancer by integrating big and diverse data of genomics.
Keywords
Target Discovery, Drug Discovery, Systems Biology, Network Medicine, Big Data of Genomics
Big Data of Genomics Profiling Cancer Subtypes and Drug Response
Along with the advance in new technologies, particularly the next generation sequencing (NGS), large-Scale genomics profiles of cancer samples are increasingly generated. With more and more large-scale genomic data becoming available, biomedical research becomes increasingly data driven or data intensive. For example, the Cancer Genome Atlas (TCGA) program [1], supported by the National Institutes of Health (NIH), has profiled over 11,000 cancer patient across 30 tumor types and subtypes. Each patient sample is profiled for the mRNA (using microarray and RNA seq), miRNA and protein expression, DNA aberrations (using DNA seq and SNP array), and the epigenomics (DNA methylation and histone modification). Integrative analyses of these data have uncovered novel subtypes of tumor and the underlying complex molecular mechanisms on various types of cancer such as breast cancer [2], squamous cell lung cancer [3,4], and Uterine cancer [5]. The International Cancer Genome Consortium (ICGC), on the other hand, provides more comprehensive genomics profiles of cancer patients in a global scale, with the goal of uncovering the genomic, transcriptomic and epigenomic changes of about 50 tumor types or subtypes [6]. All data sets in both TCGA and ICGC are publically available, which enables continued data-driven tumor investigation. The collection of the genomics data resources has been summarized in [7,8].
Genomic profiling is also conducted to unveil molecular signatures of drugs and mechanisms of drug action. In the Connectivity Map (CMAP), for example, the transcriptome signatures of 1302 small compound drugs are uncovered based on four cell lines by comparing the mRNA expression profiles before and after drug treatment [9]. The CMAP data has been used in repositioning the drugs for new indications by associating molecular signatures with reverse disease gene signatures. The CMAP datasets are further expanded by including more drugs or compounds and genetic perturbations (e.g RNAi) in the Library of Integrated Network-based Cellular Signatures (LINCS) program (http://www.lincsproject.org/). This leads to a database of gene signatures of over 5,000 compounds and 10,000 genetic perturbations on tens cell lines. Similarly, the Cancer Cell Line Encyclopedia (CCLE) [10] and Genomics of Drug Sensitivity in Cancer (GDSC) [11] also conduct extensive systematic investigations of the genomics basis of drug responses across nearly all tumor cell lines and with the database available publically.
More recently, multi-omics profiling is conducted at the single-cell level on a number of cancer types. For example, single-cell genomic sequencing has been conducted on bladder cancer [12] and breast cancer [13]. Single-cell exome sequencing has been conducted on kidney cancer [14], myeloproliferative neoplasm [15], and muscle-invasive bladder cancer [16]. Single-cell mRNA sequencing has been conducted on melanoma [17] and prostate cancer [18]. The single-cell genomic sequencing uncovers the general landscape of mutations, such as single nucleotide variations, insertions and deletion on the tumor genome. The single-cell transcriptomic sequencing reveals information about transcriptomic alterations, including those related to mRNAs, microRNAs, retained introns, alternative splicing, long-noncoding RNAs and fusion genes, with a much higher detection rate. The single-cell profiling allows the exploration of tumor heterogeneity, which is highly responsible to drug sensitivity, resistance and relapse of cancer therapy [19,20]. This further allows the development of a long-lasting therapeutic regimens of cancer by targeting tumor heterogeneity [20].
The multi-omics data of cancer is diverse and complex, and drug responses are high heterogeneous [21,22]. Thus it is challenging to integrate and interpret such big data for inferring knowledge or mechanisms and for therapeutic drug and target discovery. Among various computational approaches so far developed, the network-based approach appears to be highly promising for the genomic big data analysis.
Network-Based Approaches for Biomarker Discovery
Diseases are often regulated by a set of genes that coordinate and interact one and another in a network to maintain or regulate biological processes within a cell. This provides a basis of the network-based approach to data-driven disease biomarker discovery. For example, the human disease network and disease -gene network were investigated in [23,24]. The disease network showed the common and distinct gene functional modules of different diseases. In addition, the metabolic disease network was reconstructed in [25]. The human diseases are linked if mutated enzymes associated with them catalyze adjacent metabolic reactions [25], and the network analysis shown that the diseases with more connections to other diseases have higher prevalence and mortality rate.
Mathematically, the constraint of network can be viewed as the conditional random field (CRF), and explained as genes that functionally connected should be selected or should not be selected as biomarkers together. There are several widely used interactome databases [26-28] of protein-protein interaction or signaling networks, such as STRING [29,30], IntAct [31], MINT [32,33], BioGRID [34], Biocarta [35], Reactome [28], HPRD [26], and KEGG [27]. Such network information can serve as a constraint for selecting genes as robust biomarkers. For example, the average gene expression difference of connected genes can be used to select sub-network biomarkers for classifying the breast cancer metastasis from normal samples [36]. The discovered network biomarkers have increased reproducibility across data sets and increased classification accuracy and robustness. Moreover, gene biomarkers can be selected by the network constraint, many of which are potential disease-causal genes that regulate differentially expressed genes [37]. For example, figure 1 shows a sub-network of protein-protein interaction associated with metastatic prostate cancer. In specific, 40 genes associated with metastatic prostate cancer were obtained by using DisGeNET [38,39] online search, and then the genes were used as input of ReactomeFIPlugIn (a plugin of Cytoscape) [40,41] to to generate one sub-network (24 nodes and 46 edges) (Upper-Panel) that all nodes (out of the 40 prostate cancer genes) are directly linked (from Reactome database), and one sub-network (44 nodes (9 red nodes are linker genes; 35 green nodes are prostate cancer genes) and 147 edges) (Lower-Panel). Summaries of these signaling networks and network-based target discovery were reported in [7,42-44]. In addition, the network constraint was used in classifying cancer subtypes based on gene mutation data [45]. Individual patients often have distinct gene mutations, and it is often difficult to use the mutation data to classify patients into subtypes due to the data missing (no mutation in a given gene). To solve the problem, the mutation information of individual genes can be diffused on the protein-protein interaction network, and the clustering analysis is then conducted on the diffused mutation data to obtain meaningful cancer subtypes [45]. In addition to applied on the single type of genomics data, the network constraints is also used to integrate multiple types of genomics data, e.g., mRNA, miRNA and DNA copy number data in [46]. In brief, signaling pathways are selected as a factor graph, and the protein status is determined by the gene copy number and mRNA expression level, as well as the neighboring nodes on the factor graph [46]. It is expected that more data-driven computational methods will being developed based on integration of genomic variation data with the network constraint, contributing to the robust identification of disease related genes or signaling networks as biomarkers.
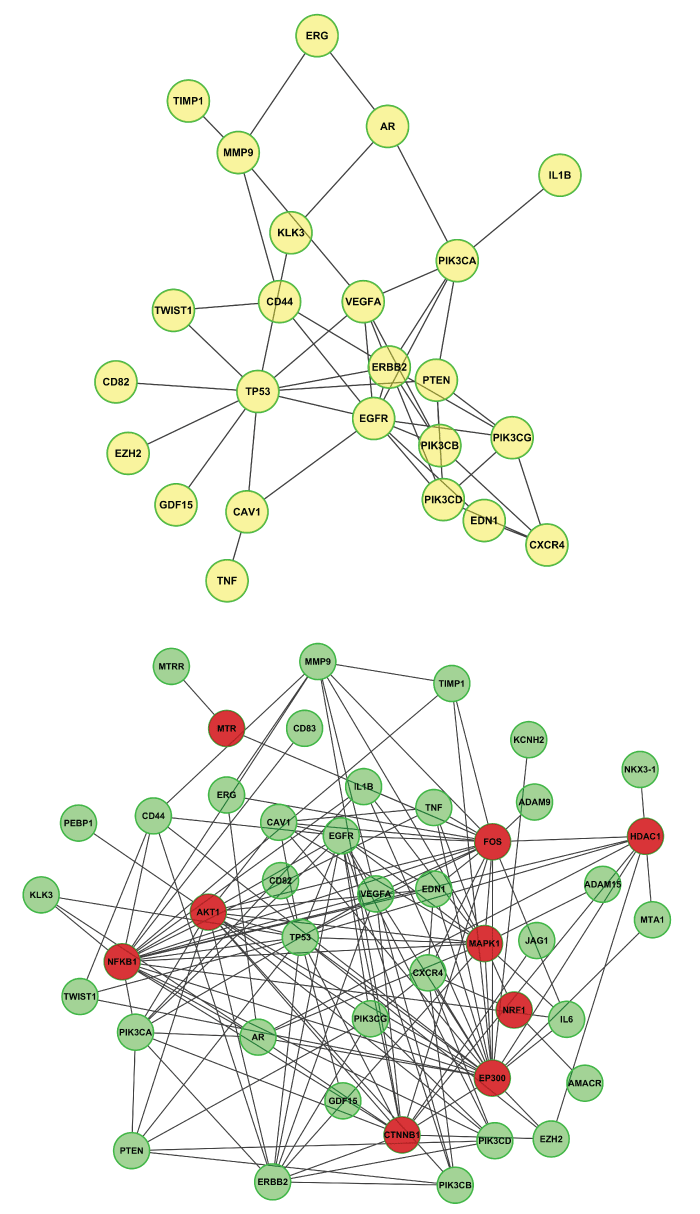
Figure 1: Examples of sub protein-protein interaction network associated with metastasis prostate cancer. Forty genes associated with metastatic prostate cancer (obtained by using DisGeNET online search), and then the genes were used as input of ReactomeFIPlugIn (a plugin of Cytoscape) to generate one sub-network (24 nodes and 46 edges) (Upper-Panel) that all nodes (out of the 40 prostate cancer genes) are directly linked (from Reactome database), and one sub-network (44 nodes (9 red nodes are linker genes; 35 green nodes are prostate cancer genes) and 147 edges) (Lower-Panel).
View Figure 1
Network-Based Approaches for Drug Discovery
Network medicine has been believed as the next paradigm of drug discovery [7,8,47]. Network-based drug discovery often employs the graph theory or topological analysis of network to visualize and identify drugs or drug combinations. For example, figure 2 shows the sub-networks of FDA approved drugs and their target proteins has shown that etiological drugs targeting disease genes have a higher odd to be effective [48]. The topological analysis of the drug-target network indicates that many drugs target on the same set of targets. (Figure 2) To expand the work, the diffusion process on the drug-target network can applied to predict unknown targets of given drugs [49]. The nearby targets of drugs that are not directly connected in the drug-target network will be potential off-targets of drugs. Moreover, the STITCH database provides a comprehensive drug-target interactions based on both know drug-target interactions as well as the literature report evidence [50].
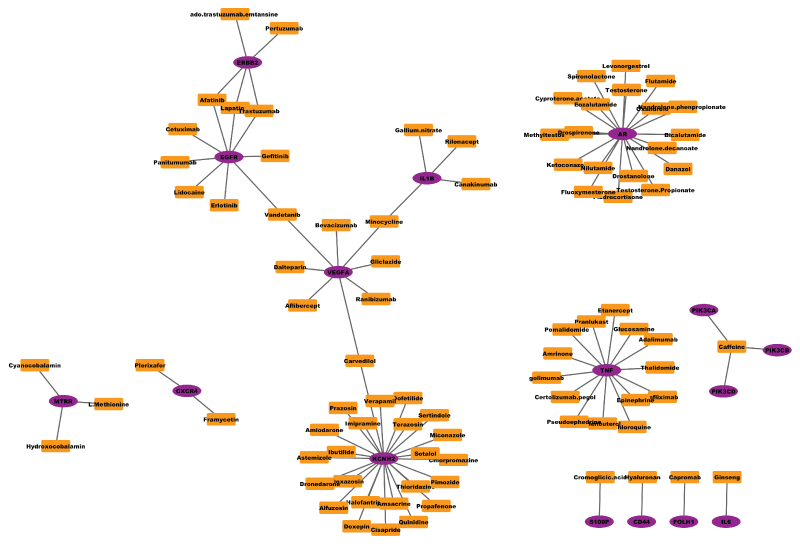
Figure 2: example drug (square shape)-target (circle shape) network. Drug-target interactions were obtained from drugbank database (Version 4.2, Released on 2015-04-01) (http://www.drugbank.ca/), and figure was plot using Cytoscape software.
View Figure 2
In addition to the drug-target network, the CMAP genomics profiling data of drugs are particularly used to reconstruct the drug-drug interaction network for drug discovery [51]. For example, similarity scores of drugs are first estimated based on the genomics data of multiple cell lines before and after drug treatment; and then the drug-drug network is constructed by linking drug pairs that have similarity scores than the given threshold [51]. Then drug-drug network is partitioned into sub drug-drug networks (or modules), in which drugs are believed share similar mechanisms of action. Based on the known targets and clinical indications of some drugs, drug targets and mechanism of other drugs in the same sub-network can be predicted. The sub drug-drug network can be also used in discovering synergistic drug combinations [50,52,53]. For example, drugs that are from different sub-networks with distinct modes of action and target on different parts of the disease signaling networks are identified and considered as have a higher degree of synergy.
Summary
Diseases are often regulated by complex signaling networks, and multi-drugs and multi-targets are often associated to form a big drug-target network. The network-based approach is thus a logic choice for the data-driven therapeutic discovery. Topological structure of networks provides constraints to select more robust and causal network biomarkers, associates drugs with the targets through the information diffusion process. An increasing number of network-based approaches have been developed for biomarker and drugs discovery. The development is particularly benefited from the availability of many well established bionetwork analysis and visualization tools (e.g., igraph in R (http://igraph.org/r/), NetworkX in Python (https://networkx.github.io/), Cytoscape (http://www.cytoscape.org/)). Yet, challenges remain in discovering informative biomarkers and effective drugs. More sophisticated or comprehensive network-based approaches to therapeutic discovery are expected in the new era of pharmacogenomics research.
References
-
Zhu H, Han C, Wu T (2015) MiR-17-92 cluster promotes hepatocarcinogenesis. Carcinogenesis 36: 1213-1222.
-
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61-70.
-
Cancer Genome Atlas Research Network (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519-525.
-
Wilkerson MD, Yin X, Hoadley KA, Liu Y, Hayward MC, et al. (2010) Lung squamous cell carcinoma mRNA expression subtypes are reproducible, clinically important, and correspond to normal cell types. Clin Cancer Res 16: 4864-4875.
-
Cancer Genome Atlas Research Network, Kandoth C, Schultz N, Cherniack AD, Akbani R, et al. (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497: 67-73.
-
https://icgc.org/
-
Barabasi AL, Gulbahce N, Loscalzo J (2011) Network medicine: a network-based approach to human disease. Nat Rev Genet 12: 56-68.
-
Werner HM, Mills GB, Ram PT (2014) Cancer Systems Biology: a peek into the future of patient care? Nat Rev Clin Oncol 11: 167-176.
-
Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, et al. (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929-1935.
-
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, et al. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603-607.
-
Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, et al. (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570-575.
-
Morrison CD, Liu P, Woloszynska-Read A, Zhang J, Luo W, et al. (2014) Whole-genome sequencing identifies genomic heterogeneity at a nucleotide and chromosomal level in bladder cancer. Proc Natl Acad Sci U S A 111: E672-681.
-
Navin N, Kendall J, Troge J, Andrews P, Rodgers L, et al. (2011) Tumour evolution inferred by single-cell sequencing. Nature 472: 90-94.
-
Xu X, Hou Y, Yin X, Bao L, Tang A, et al. (2012) Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell 148: 886-895.
-
Hou Y, Song L, Zhu P, Zhang B, Tao Y, et al. (2012) Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148: 873-885.
-
Li Y, Xu X, Song L, Hou Y, Li Z, et al. (2012) Single-cell sequencing analysis characterizes common and cell-lineage-specific mutations in a muscle-invasive bladder cancer. Gigascience 1: 12.
-
Ramskold D, Luo S, Wang YC, Li R, Deng Q, et al. (2012) Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30: 777-782.
-
Welty CJ, Coleman I, Coleman R, Lakely B, Xia J, et al. (2013) Single cell transcriptomic analysis of prostate cancer cells. BMC Mol Biol 14: 6.
-
Castells M, Thibault B, Delord JP, Couderc B (2012) Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci 13: 9545-9571.
-
Somasundaram R, Villanueva J, Herlyn M (2012) Intratumoral heterogeneity as a therapy resistance mechanism: role of melanoma subpopulations. Adv Pharmacol 65: 335-359.
-
Zlotta AR (2013) Words of wisdom: Re: Genome sequencing identifies a basis for everolimus sensitivity. Eur Urol 64: 516.
-
Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, et al. (2012) Genome sequencing identifies a basis for everolimus sensitivity. Science 338: 221.
-
Midic U, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN (2009) Protein disorder in the human diseasome: unfoldomics of human genetic diseases. BMC Genomics 10: S12.
-
Hu G, Agarwal P (2009) Human disease-drug network based on genomic expression profiles. PLoS One 4: e6536.
-
Lee DS, Park J, Kay KA, Christakis NA, Oltvai ZN, et al. (2008) The implications of human metabolic network topology for disease comorbidity. Proc Natl Acad Sci USA 105: 9880-9885.
-
Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. (2009) Human Protein Reference Database--2009 update. Nucleic Acids Res 37: D767-772.
-
Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, et al. (2008) KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480-484.
-
Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, et al. (2009) Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res 37: D619-622.
-
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, et al. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39: D561-568.
-
von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, et al. (2003) STRING: a database of predicted functional associations between proteins. Nucleic Acids Res 31: 258-261.
-
Hermjakob H, Montecchi-Palazzi L, Lewington C, Mudali S, Kerrien S, et al. (2004) IntAct: an open source molecular interaction database. Nucleic Acids Res 32: D452-455.
-
Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, et al. (2002) MINT: a Molecular INTeraction database. FEBS Lett 513: 135-140.
-
Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, et al. (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 40: D857-861.
-
Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, et al. (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Res 34: D535-539.
-
http://www.biocarta.com/
-
Chuang HY, Lee E, Liu YT, Lee D, Ideker T (2007) Network-based classification of breast cancer metastasis. Mol Syst Biol 3: 140.
-
Navlakha S, Kingsford C (2010) The power of protein interaction networks for associating genes with diseases. Bioinformatics 26: 1057-1063.
-
Pinero J, Queralt-Rosinach N, Bravo A, Deu-Pons J, Bauer-Mehren A1, et al. (2015) DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford) 2015: bav028.
-
Bauer-Mehren A, Rautschka M, Sanz F, Furlong LI (2010) DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics 26: 2924-2926.
-
Wu G, Dawson E, Duong A, Haw R, Stein L (1000) ReactomeFIViz: a Cytoscape app for pathway and network-based data analysis. Version 2.
-
Wu G, Feng X, Stein L (2010) A human functional protein interaction network and its application to cancer data analysis. Genome Biol 11: R53.
-
Prieto C, De Las Rivas J (2006) APID: Agile Protein Interaction DataAnalyzer. Nucleic Acids Res 34: W298-302.
-
Calderone A, Castagnoli L, Cesareni G (2013) mentha: a resource for browsing integrated protein-interaction networks. Nat Methods 10: 690-691.
-
Wist AD, Berger SI, Iyengar R (2009) Systems pharmacology and genome medicine: a future perspective. Genome Med 1: 11.
-
Hofree M, Shen JP, Carter H, Gross A, Ideker T (2013) Network-based stratification of tumor mutations. Nat Methods 10: 1108-1115.
-
Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, et al. (2010) Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26: 237-245.
-
Pujol A, Mosca R, Farres J, Aloy P (2010) Unveiling the role of network and systems biology in drug discovery. Trends Pharmacol Sci 31: 115-123.
-
Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M (2007) Drug-target network. Nat Biotechnol 25: 1119-1126.
-
Cheng F, Liu C, Jiang J, Lu W, Li W, et al. (2012) Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput Biol 8: e1002503.
-
Kuhn M, Szklarczyk D, Franceschini A, von Mering C, Jensen LJ, et al. (2012) STITCH 3: zooming in on protein-chemical interactions. Nucleic Acids Res 40: D876-880.
-
Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, et al. (2010) Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A 107: 14621-14626.
-
Huang L, Li F, Sheng J, Xia X, Ma J, et al. (2014) DrugComboRanker: drug combination discovery based on target network analysis. Bioinformatics 30: 228-236.
-
Lee JH, Kim DG, Bae TJ, Rho K, Kim JT, et al. (2012) CDA: combinatorial drug discovery using transcriptional response modules. PLoS One 7: e42573.
