Residual feed intake (RFI) is the difference between actual and predicted feed intake and an important factor determining feed efficiency (FE). Recently, 170 candidate genes were associated with RFI, but no expression quantitative trait loci (eQTL) mapping has hitherto been performed on FE related genes in dairy cows. In this study, an integrative systems genetics approach was applied to map eQTLs in Holstein and Jersey cows fed two different diets to improve identification of candidate genes for FE.
Liver RNA-seq transcriptomics data from nine Holstein and ten Jersey cows that had been fed control (C) or high concentrate (HC) diets were integrated with genomic data (from 777k BovineHD Illumina BeadChip) by using the Matrix eQTL R package. A total of 170 previously identified candidate genes for FE (89 differentially expressed genes (DEGs) between high and low RFI groups and 81 hub genes (HG) in a group of co-expressed genes) were used in the data integration analysis.
From the 241,542 SNPs used in the analysis, we identified 20 significant (FDR < 0.05) local-eQTLs targeting seven candidate genes and 16 significant (FDR < 0.05) local-eQTLs targeting five candidate genes related to RFI for the C and HC diet group analysis, respectively, in a breed-specific way.
Interestingly, Holstein and Jersey cows appear to rely on different strategies (lipid and cholesterol metabolism versus immune and inflammatory function) to achieve low RFI. The eQTLs overlapped with QTLs previously associated with FE trait (e.g. dry matter intake, longevity, body weight gain and net merit). The eQTLs and biological pathways identified in this study improve our understanding of the complex biological and genetic mechanisms that determine FE traits in dairy cattle. The identified eQTLs/genetic variants can potentially be used in new genomic selection methods that include biological/functional information on SNPs.
eQTL, RNA-seq, Genotype, Data integration, Systems genomics, Feed efficiency, Residual feed intake
ANOVA: Analysis of Variance; C: Low Concentrate (Control); DCRC: Danish Cattle Research Centre; DEGs: Differentially Expressed Genes; EDTA: Ethylenediaminetetraacetic Acid; eQTL: Expression Quantitative Trait Loci; FDR: False Discovery Rate; FE: Feed Efficiency; HC: High Concentrate; HG: Hub Genes; Mb: Mega Base; QTL: Quantitative Trait Loci; RFI: Residual Feed Intake; RNA: Ribonucleic Acid; RNA-seq: RNA Sequencing; SNPs: Single Nucleotide Polymorphisms; WGCNA: Weighted Gene Co-expression Analysis.
Feed intake and the conversion of absorbed nutrients into milk components are major determinants of feed efficiency (FE) in dairy cattle and hence production economics. FE is a complex trait that is influenced by several genetic and environmental factors, which in an interactive way control feed intake, nutrient partitioning and metabolic adaptation to lactation in different body tissues as well as milk synthesis and immune function. In dairy cattle, the use of FE for breeding purposes is quite complicated, since recording of individual feed intake is difficult in group fed cows. It is therefore desirable to be able to predict the genetic contributions to this trait to be able to select the most feed efficient cows for breeding purposes.
To date, transcriptomics has given precise and reliable results that identify candidate genes related to phenotypes of interest [1]. Although gene expressions associated with FE related genes have been studied for a long time, also in cattle [2-4], genetic markers are more easily accessible and not affected by environmental factors in contrast to gene expression data.
However, in some cows, the actual feed intake deviate from the predicted by their genetic heritage, even when they are exposed to similar environmental conditions. The term residual feed intake (RFI) describes this deviation and is calculated as the difference between the actual measured and the predicted feed intake of the cow [5]. Among groups of high and low RFI cattle, we have recently identified several candidate genes that predict the RFI in Danish dairy cattle [6].
Therefore, in this present study we focused on genetic markers for RFI in an attempt to improve the prediction of genetic merit for FE, which is needed to be able to use this type of determinants in practice.
Integration of transcriptomics and genomics data can be used to identify potential causal genetic variants that affect particular phenotypes. This approach is known as Genetical Genomics or Integrative Genomics [7]. The identified regions are called expression Quantitative Trait Loci (eQTL). In other words, an eQTL is a region in a particular locus that influences or controls the differences of expressions of causal genes [8-11]. The expression profile is an intermediate biological space between the phenotype and the genome. Therefore, eQTL analysis can identify interesting genetic variants even with a low sample size [12,13]. The identification of genomic regions influencing the expressions of the candidate genes could give a better perspective to use the information in animal selection as well as provide a better explanation about the way genomic regions control traits of interest.
A few studies have been conducted to identify genomic regions determining FE traits in beef cattle, chicken and other livestock species [14-17]. However, no eQTL mapping has hitherto been performed on FE related genes in dairy cows.
In this study, we performed an eQTL mapping analysis on candidate genes for the RFI trait. The hypothesis of the integrative genomics analysis is that SNPs associated with the expression of candidate genes are involved or in linkage with genomic regions regulating their expression. Therefore, the objective of this study was to identify eQTL regions together with their functional annotations associated with the RFI trait in two breeds of dairy cattle (Danish Holstein and Danish Jersey) fed two different diets and to present an eQTL mapping of candidate genes for RFI using matrix eQTL analysis, as well as characterize the SNPs by comparing our findings with previously annotated QTLs. The eQTL identified in this study could be important candidate genetic markers defining actual FE in dairy cattle, and our study suggests that there are differential traits relating to RFI in Danish Holsteins as compared to Jerseys.
The present study is based on biological samples obtained from nine Holstein and ten Jersey cows, housed at the Danish Cattle Research Centre (DCRC), Aarhus University, Denmark. The animals were part of a larger experiment, where FE was determined in 200 dairy cows distributed on the two breeds [18]. The details about the animal's background and the overall experimental design of the larger trial can be found in Salleh, et al. and Li, et al. [6,18].
The experimental cows used in the present study were selected based on individually recorded RFI of cows from the larger study. A total of four Holstein cows with very high and five with very low RFI, and five Jersey cows with very high and five with very low RFI were selected, and their deviation from the average recorded RFI is shown in Figure 1. The experimental cows underwent two periods of feeding trials low concentrate (control (C)) and high concentrate (HC) diet. The two dietary exposures were separated by a conditioning period of 14 - 26 days. The details of the ration composition for both diet can be found in Salleh, et al. [6].
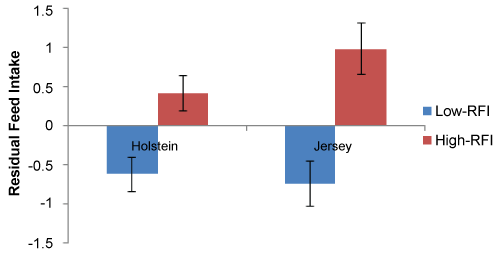 Figure 1: Mean ± SE of Residual Feed Intake (RFI) value for the Holstein and Jersey cows used in the present experiment [6].
View Figure 1
Figure 1: Mean ± SE of Residual Feed Intake (RFI) value for the Holstein and Jersey cows used in the present experiment [6].
View Figure 1
Liver biopsies (approximately 20 mg) were collected from each cow at the end of each feeding trial, RNA was extracted and sequenced. The details of the samples collection and processing were described in Salleh, et al. [6].
Blood samples were used for the DNA genotyping procedure. Ten milliliters blood samples were collected from the 19 cows using Ethylenediaminetetraacetic acid (EDTA) coated blood tubes. The blood samples were stored at -20 ℃ pending genomic DNA isolation and genotyping. The DNA was isolated and genotyped by Neogen GeneSeek® (Lincoln, NE, USA) using 777k BovineHD BeadChip (Illumina, Inc., San Diego, CA, USA).
Briefly, the RNA-seq data were pre-processed and processed to find candidate gene through differential expression analysis and weighted gene co-expression network analysis (WGCNA). RNA-seq analysis was performed as previously described in Salleh, et al. [6]. Briefly, the RNA raw reads were pre-processed using FastQC version 0.11.3 [19]. The reads were aligned to the Bovine reference genome release 82 using STAR aligner [20]. After the alignment, quality control of the mapped reads was done using Qualimap version 2.0 [21]. Then the HTSeq-count tool was used to compute the gene expression counts [22]. The DEGs analyses were done using DESeq2 package [23] and weighted gene co-expression analyses using WGCNA package [24]. Hub genes were selected from the top significant modules that have significant association with RFI trait and having more than 80% module membership. The RNA-seq data for the present study is available in http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92398.
Subsequently, the two gene expression datasets of Holsteins and Jerseys were preprocessed independently to filter low counts genes in each breed. Next, the two datasets were merged by keeping only genes present in both RNA-seq datasets.
We performed two separate analyses as replicates, one using the expression profile of the cows on C diet and another one using the expression profile of the cows on the HC diet. A summary of the eQTL mapping pipeline is presented in Figure 2. The eQTL mapping analysis was performed on 170 candidate genes for RFI (Supplementary Table 1, Supplementary Table 2, Supplementary Table 3 and Supplementary Table 4) that were identified in two previous studies based on the same RNA-seq data. The total list of candidate genes included 89 DEGs between cows with high and low RFI [6] and 81 hub genes in groups of co-expressed genes associated with RFI identified by using a weighted gene co-expression network analysis (WGCNA) (unpublished).
 Figure 2: eQTL mapping pipeline used to find candidate genes for Residual Feed Intake (RFI) in dairy cattle. Among 170 candidate genes, only the 160 genes present in RNA-seq datasets for both breeds were used in the further analyses.
View Figure 2
Figure 2: eQTL mapping pipeline used to find candidate genes for Residual Feed Intake (RFI) in dairy cattle. Among 170 candidate genes, only the 160 genes present in RNA-seq datasets for both breeds were used in the further analyses.
View Figure 2
Table 1: Top significant local-eQTLs targeting candidate genes for cows fed the control (C) diet. View Table 1
Table 2: Top significant local-eQTLs targeting candidate genes for cows fed the high concentrate (HC) diet. View Table 2
Among the 170 candidate genes, 160 survived after the filtering step in both datasets and were used in the rest of the analyses. The numbers of candidate genes that survived after filtration were the same in the two separate analyses, which were performed for cows fed low as compared to high concentrate diets. The log2 transformation of the gene count matrix was used in the eQTL mapping.
The genotype data was filtered by Hardy Weinberg Equilibrium (HWE < 0.0001), Minor Allele Frequency (MAF < 0.15), and missing genotype rates (mind > 0.1). The genotype data were also pruned to remove SNPs in strong linkage disequilibrium [25]. The preprocessing was performed using PLINK 1.90 beta software [26]. A total of 536,420 SNPs was removed after the filtration procedure. The remaining 241,542 SNPs were used for the rest of the analysis.
The theoretical aspects of eQTL mapping and applications of findings in animal sciences are well described in the literature, including our previous studies [9-11]. The eQTL mapping was performed by fitting an analysis of variance (ANOVA) model to test both additive and dominant effects. The Matrix eQTL v2.1.1 [27] package in R software was used to identify the local and distant-eQTL associated to the RFI trait. We included the breed and the lactation number as covariates in the model.
The local-and distant-eQTLs analyses were performed separately. The analysis of local eQTL was performed on SNPs that were located at less than 1 Mb distance from the start or end position of the gene of interest, while distant-eQTLs analysis was performed on SNPs located at a distance of more than 1 Mb on the same chromosome and on SNPs in other chromosomes. The SNPs were mapped onto the Bos taurus genome UMD 3.1. The information about gene locations were retrieved from Ensembl database for Bos taurus v82. P-values were adjusted using the false discovery rate (FDR) procedure for multiple comparison corrections [28]. SNPs were considered significant with FDR lower than 0.05.
The significant eQTLs identified in this study were further compared to the Animal Genome cattle QTLdb database [29]. From the cattle QTL database we filtered out long QTL regions and more than one flank markers. In total, 94,322 SNPs were used in the comparison. The SNPs information was obtained from 337 studies, 63 breeds, 366 traits of 6 trait types. The flanking regions of 500 kb around the eQTL identified in our studies were compared against the cattle QTLdb. The QTLs overlapping for at least one nucleotide were considered as a match.
The eQTL mapping analysis allows identification of SNPs associated with the expression level of specific genes. The hypothesis of this analysis is that the eQTL or eSNPs are in linkage with regulatory regions or region that encode for transcription factors responsible for the control of the expression of the targeted gene [10]. In the present study, we have analyzed candidate genes associated with the RFI trait in dairy cattle. Despite the small sample size, we identified several loci significantly related to the expression of the candidate genes. In addition, since the study focused on genes significantly associated with RFI, and the eQTL analysis was done on animals with widely different RFI, either very low or very high, our study had enough power to detect biologically meaningful expression variants.
In the expression profile of cows fed the control diet, we identified 20 local-eQTLs SNPs or cis-eQTLs SNP (FDR < 0.05) associated with the expression of seven genes (BDH2, CHRNE, ELOVL6, GIMAP4, FDXR, CXCL9 and CD52) (Table 1). However, there was no significant distant-eQTL (trans-eQTLs) associated with the candidate genes in the analysis performed among cows fed the control diet. On the other hand, among cows fed the high concentrate diet, we identified 16 local eQTLs SNPs (FDR < 0.05) associated with the expression of five genes (UHRF1, BDH2, HSD17B4, GIMAP4 and ENSBTAG00000047529) (Table 2) and 2891 distant-eQTLs associated with the expression of 45 genes. Among the local-eQTL, genes that were in common in both diet groups were the BDH2 and GIMAP4 genes. Figure 3 shows the significant eQTLs targeting candidate genes. A complete list of the significant distant-eQTLs, including the chromosomal position and annotation of the SNPs in the HC dataset analysis is presented in Supplementary Table 5.
 Figure 3: Venn diagram showing the participation of the significant eQTLs targeting candidate genes in cows when fed control (C) as compared to high concentrate (HC) diets.
View Figure 3
Figure 3: Venn diagram showing the participation of the significant eQTLs targeting candidate genes in cows when fed control (C) as compared to high concentrate (HC) diets.
View Figure 3
In Holstein, cows fed with C diet dataset, we detected eQTLs associated to the three genes BDH2, CHRNE and ELOVL6, whereas for cows fed the HC diet dataset, the eQTLs associated to two other genes, UHRF1 and HSD17B4. In Jersey cows, two DEGs (GIMAP4 and FDXR) and two HG's (CXCL9 and CD52) belonging to a group of co-expressed genes associated with RFI, when they were fed the C diet, whereas only the GIMAP4 gene was detected as significant local-eQTL, when they were fed the HC diet. However, the HG's (CXCL9 and CD52) were not further analyzed, since only one animal with rare allele at these loci were present in the dataset. Supplementary Figure 1 and Supplementary Figure 2 present boxplots of genotypes and their correlation with gene expressions for the top seven significant local-eQTLs corresponding to seven candidate genes (BDH2, CHRNE, ELOVL6, GIMAP4, FDXR, UHRF1 and HSD17B4) for the RFI trait in the present study.
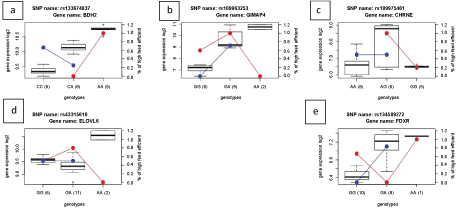 Supplementary Figure 1: a-e) The boxplots show the five significant eQTLs with the associated genes for control diet group analysis. X-axis: genotypes; y-axis: gene expression (log2); red line: Holstein; blue line: Jersey.
View Supplementary Figure 1
Supplementary Figure 1: a-e) The boxplots show the five significant eQTLs with the associated genes for control diet group analysis. X-axis: genotypes; y-axis: gene expression (log2); red line: Holstein; blue line: Jersey.
View Supplementary Figure 1
 Supplementary Figure 2: The boxplots show the two significant eQTLs with the associated genes for high concentrate diet group analysis. x-axis: genotypes; y-axis: gene expression (log2); red line: Holstein; blue line: Jersey.
View Supplementary Figure 2
Supplementary Figure 2: The boxplots show the two significant eQTLs with the associated genes for high concentrate diet group analysis. x-axis: genotypes; y-axis: gene expression (log2); red line: Holstein; blue line: Jersey.
View Supplementary Figure 2
Previous studies showed that identification of eQTLs and genomic regions would give additional information towards the identification of causal variants [30]. Hence, the eQTLs that were identified as associated to the RFI trait in the present study would provide additional information for the development of biomarkers.
The first top eQTL with a significant relationship between the gene expression and genotype is rs133674837, which is associated to the BDH2 gene (Supplementary Figure 1a), and as mentioned the association was found to be significant in the two separate analyses for cows when fed the C diet as well as when fed the HC diet. The expression of the BDH2 gene was previously identified to be upregulated in high FE cows [6]. All low RFI (high FE) Holstein cows (n = 5) had homozygous (AA) genotype at this locus, while 80% of the high RFI (low FE) Holstein cow had heterozygous genotype (CA). BDH2 encodes for the enzyme 3-Hydroxybutyrate Dehydrogenase 2, which is responsible for degradation of 3-hydroxybutyrate-a ketone body derived partly from rumen fermentation and partly from incomplete oxidation of fatty acids in the liver [31,32]. The BDH2 gene in the liver has been observed to be downregulated in animals, when ketogenesis occurred (mice and pigs) [31,33]. This happens, for example, during feed restriction or fasting of animals, and mRNA expression of BDH2 gene has been shown to be lower in such animals compared to normal feeding animals [33]. In the present study, the hepatic BDH2 gene expression was downregulated in high RFI (low FE) animals. The positive association between a homozygous (AA) genotype and upregulation of the BDH2 gene in low RFI (high FE) Holstein cows shows that this locus might influence the RFI trait. However, in Jersey cows, 80% (n = 4) of low RFI (high FE) cows were homozygous (CC) at this allele. Hence, specifically for Holstein cows, a homozygous (AA) genotype is expected to favor low RFI and hence high FE.
GIMAP4 gene is another gene that has been detected as significantly associated with the eQTLs listed in Table 1 and Table 2 in both analysis (i.e. when cows were fed the C and HC diets). The top significant eQTLs targeting GIMAP4 was rs109963253. All the five low RFI (high FE) Jersey cows were heterozygous (GA) at this SNP locus, while 60% (n = 3) of the high RFI (low FE) cows were homozygous (GG) (Supplementary Figure 1b). GIMAP4 encodes for a GTPase binding protein responsible for regulating lymphocyte apoptosis (http://www.genecards.org/cgi-bin/carddisp.pl?gene=GIMAP4). Hence in humans, the GIMAP4 gene has been shown to be involved in immunological responses [34]. We already in our previous paper [6] stated the importance of the GIMAP genes for the FE trait, since the GIMAP4 gene was significantly higher expressed in high feed efficient than low feed efficient Jersey cows. The present study is thus in line with conclusions from previous studies, where function of immunological responses has been associated to productivity and FE in farm animals [35-38]. A possible explanation is that animals with poorer immune function are more prone to develop infectious diseases like e.g. mastitis, and this can reduce milk production, induce fever-associated increases in metabolism, and hence increased energy expenditures per kg of produced milk, which subsequently reduces FE [39].
rs109975461, which is associated with the CHRNE gene, was also a significant eQTL. At this locus, all high RFI (low FE) Holstein cows had a homozygous (GG) genotype, whereas 80% of the low RFI (high FE) cows (n = 4) had a heterozygous (AG) genotype (Supplementary Figure 1c). In other words, high feed efficient cows that had a high expression of the CHRNE gene also had the heterozygote genotype. However, in the Jersey group, there was no association to be seen for this CHRNE gene. The CHRNE gene encodes for the acetylcholine receptors in mature mammalian neuromuscular junctions. In general, this gene was never discussed before in relation with FE traits. Acetylcholine has been reported to influence hepatocyte glucose metabolism in rodents via actions on muscarinic receptors [40], but whether this is also the case in ruminants is not clear. Perhaps, more importantly, acetylcholine plays a critical role in the complex regulation of hypothalamic neuronal activity that influences feed intake [41], and in dairy cows, feed intake is a major factor limiting milk production in high-yielding dairy cows in early lactation [42].
Another interesting candidate identified in the analyses of Holstein cows on the C diet was the ELOVL6 gene. In our study, the top SNP targeting ELOVL6 gene was rs43315610. The ELOVL6 gene has previously been discovered as an important gene that influences FE in beef cattle and pigs [43,44]. ELOVL6, which is also known as elongation of very long chain fatty acids protein 6, is part of the pathway of de novo fatty acid synthesis [45]. The lower expression of this gene in low RFI Holstein cows might be associated with low rates of de novo synthesis of fatty acids, as it has previously been described in pigs [44,46], and de novo synthesized fatty acids constitute up to 50% of fatty acids in milk on a molecular weight basis [47]. This gene has also been associated to long chain fatty acid synthesis in beef cattle [48]. Previously, the expression of ELOVL6 was found differentially expressed in liver, adipose tissue and muscle [48]. In another study on QTL mapping for RFI in Holstein calves, it was found that another gene involved in fatty acid metabolism, FABP4 gene were significantly associated with the top SNPs significantly associated RFI across three stages of age [49]. This gene is encoded for fatty acid binding protein which suggests that fatty acid synthesis and metabolism may be important parts of the RFI trait. In Jersey cattle, we did not observe any relation between RFI genotype and the ELOVL6 gene expression. We found that 80% (n = 4) of the low RFI (high FE) Holstein cows had a heterozygous (GA) genotype, while 20% (n = 1) were homozygous (GG) (Supplementary Figure 1d). Therefore, a heterozygous genotype is expected to favor high FE.
When cows were fed the C diet, rs134589272 was identified as an eQTL, which corresponded to the FDXR gene in Jersey cows. All (n = 5) low RFI (high FE) Jersey cows were heterozygous (GA) and had high expression of this gene, while 80% (n = 4) of the high RFI (low FE) Jersey cows were homozygous (GG) corresponding to a lower expression of the FDXR gene (Supplementary Figure 1e). For Holstein cows, RFI was not related neither to this eQTL nor to the genotype for the FDXR gene. Functions of the FDXR gene are related to cholesterol metabolism [6], which is an important feature of e.g. membrane synthesis, which is important for formation of the milk fat globule membrane covering secreted milk fat.
In addition, another four eQTLs associated to the gene HSD17B4 and UHRF1 expression were found as significant in the analysis for cows fed the HC diet (Supplementary Figure 2a and Supplementary Figure 2b). Interestingly, these two genes were also previously found associated with the FE trait. The HSD17B4 gene encodes for a major enzyme involved in peroxisomal β-oxidation, and it was found to be upregulated in abdominal fat of low growth chicken [50], and this appears to be in line with the present study, where the HSD17B4 gene expression was upregulated in the high RFI Holstein cows. UHRF1 encodes for Ubiquitin like With PHD and Ring Finger Domains 1 (http://www.genecards.org/cgi-bin/carddisp.pl?gene=UHRF1), which is an essential regulator of DNA methylation. Several studies have identified that Ubiquitin family genes were significantly associated with RFI traits in Bos taurus [51,52]. In the present study UHRF1 gene was found significantly downregulated in high RFI Holstein cows.
In order to gain more information regarding the eQTLs that we discovered in the present study, we compared the results of the SNPs locations with the previously reported QTLs and variants from GWAS study from the Animal genome cattle QTL database. We identified several overlaps of our eQTLs with QTLs from previous studies. The QTLs overlapping with our eQTLs were associated with a different type of traits (Figure 4a and Figure 4b).
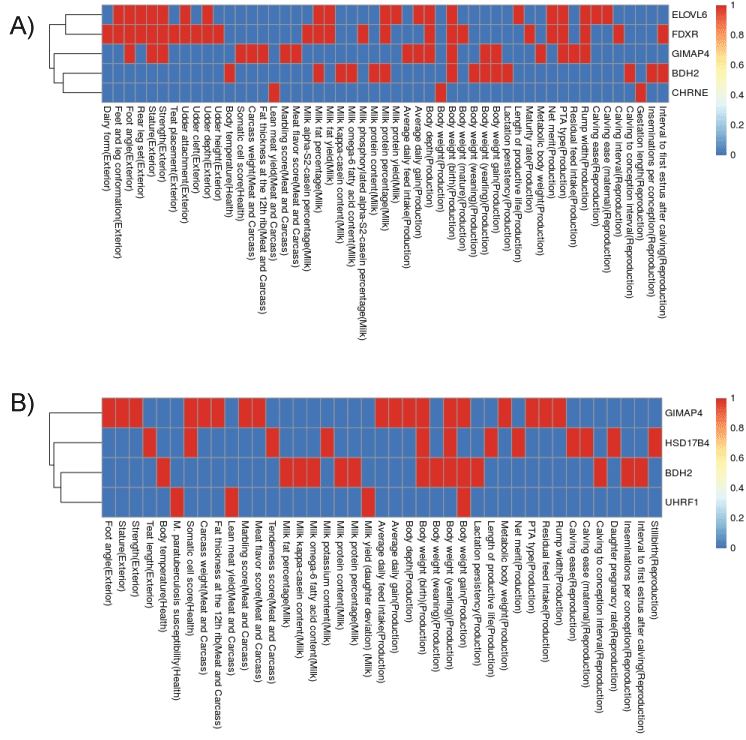 Figure 4: Heatmap showing SNPs corresponding to seven genes overlapping with previous QTLs for major traits in cattle QTL database (a) Local-eQTLs associated with genes in cows on the control (C) diet analysis; (b) Local-eQTLs associated with genes in cows on the hogh concentrate (HC) diet. Red: with at least one hit; Blue: no hit.
View Figure 4
Figure 4: Heatmap showing SNPs corresponding to seven genes overlapping with previous QTLs for major traits in cattle QTL database (a) Local-eQTLs associated with genes in cows on the control (C) diet analysis; (b) Local-eQTLs associated with genes in cows on the hogh concentrate (HC) diet. Red: with at least one hit; Blue: no hit.
View Figure 4
The eQTLs which associated to the expression of ELOVL6 and FDXR genes are the most overlapped with many traits. Only the GIMAP4 gene was previously associated to production traits, such as RFI, rump width, metabolic body weight, body weight gain, body weight (yearling), body weight, body depth, average daily gain as well as average daily feed intake [53,54]. However, the same region contains QTLs for other traits, such as reproduction, milk, meat and carcass, health and exterior association traits [55,56].
The fact that all these associations with different type of traits were found within this 1Mb region, shows that this must be a significant region with control points for several targeting genes. The eQTLs identified are close to the QTL for production traits and for FE traits. At the same time, this confirms that the candidate genes which associated to FE trait in our findings were also closely associated to several production traits. However, the association with other important traits can be a sign of double association between reproduction and production traits, which were well discussed elsewhere [57]. Thus, the uses of genomic region information need to be tested and validated in a different and a larger population before further usage in any genomic selection procedures can be implemented.
To bridge the gap between genotype and phenotype, we attempted in this study to identify DEGs and HG's among previously identified candidate genes for the FE trait. The identified local-eQTLs provide additional evidence of the involvement of some of previously identified candidate genes in RFI determination, and our study provides new information on possible regulatory and causative genetic variants that can be used in genomics-based selection for FE in dairy cows. We identified eQTL associated to the expression of seven genes (BDH2, CHRNE, ELOVL6, GIMAP4, UHRF1, HSD17B4 and FDXR) that appear to be involved in metabolic pathways related to RFI and hence feed efficiency. The eQTLs overlapped with QTLs previously associated with FE trait (e.g. dry matter intake, longevity, body weight gain and net merit). Interestingly, Holstein and Jersey cows appear to rely on different strategies to achieve low RFI, and this was associated to cholesterol and lipid metabolism related pathways in Holstein cows, but to immune and inflammatory related functions in Jersey cows. Thus, our findings suggest that the identified eQTLs can be used as potential biomarkers for feed efficiency and used to predict feed efficiency level. The genomic region around the identified SNP markers could be included in genomics/genetic-based selection in Holstein and Jersey. However, before applying this new knowledge in genetic testing or in commercial applications, the results must be validated in a larger population, and it must be further analyzed if pleiotropic effects of eQTLs also include adverse disease traits.
The authors would like to thank the collaborator scientists, specifically Peter Lund, Johanna Höglund and Dana Olijhoek, for contributions to the present study. This project was funded by Milk Levy Foundation, Skejby Denmark. The PhD student (Suraya Mohamad Salleh) would also like to acknowledge the Ministry of Higher Education of Malaysia and Universiti Putra Malaysia for the PhD stipend as well as University of Copenhagen for the tuition fee waiver.