

Journal of Rheumatic Diseases and Treatment
A Rare Coexistence of Seronegative Enthesopathy and Arthropathy Syndrome with Familial Mediterranean Fever
Hakan Genç1*, Alper Gümüstepe1 , Fikriye Figen Ayhan1,2 and Aynur Karagöz1
1Department of Physical Medicine and Rehabilitation, Ministry of Health, Ankara Training and Research Hospital, Turkey
2Department of Physical Medicine and Rehabilitation, Usak University, High School of Health Sciences, Turkey
*Corresponding author: Hakan Genç, Physical Medicine and Rehabilitation Clinic, Ankara Training and Research Hospital, Ulucanlar Street, 06030 Altindag/Ankara, Turkey, Tel: 312 5953405, E-mail: hakangenc06@hotmail.com
J Rheum Dis Treat, JRDT-1-026, (Volume 1, Issue 4), Case Report; ISSN: 2469-5726
Received: October 20, 2015 | Accepted: November 16, 2015 | Published: November 18, 2015
Citation: Genç H, Gümüstepe A, Ayhan FF, Karagöz A (2015) A Rare Coexistence of Seronegative Enthesopathy and Arthropathy Syndrome with Familial Mediterranean Fever. J Rheum Dis Treat 1:026. 10.23937/2469-5726/1510026
Copyright: © 2015 Genç H, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Several studies showed an increase in the frequency of MEFV mutations among children with vasculitic and rheumatic diseases. Association between seronegative enthesopathy and arthropathy syndrome, and familial mediterranean fever has not been reported previously. The present case report may therefore be of interest.
Keywords
FMF, SEA syndrome, Genetics, MEFV mutations
Introduction
The juvenile-onset spondyloarthropathies begin at age of 16 years or younger and are associated with the HLA-B27 allele. They are classified as the enthesitis-related arthritis (ERA) subgroup of juvenile idiopathic arthritis (JIA) by the international league of associations for rheumatology (ILAR) [1]. Although the adult form has inflammatory low back pain as the predominant clinical symptom, the juvenile form has peripheral enthesitis and arthritis as its main clinical features. For this reason, Rosenberg and Petty proposed the concept of the seronegative enthesopathy and arthropathy (SEA) syndrome. They define this syndrome as enthesitis with arthralgia or arthritis in children less than 17 years of age who also lack rheumatoid factor (RF) and antinuclear antibodies [2].
Familial Mediterranean Fever (FMF) is a genetic disorder associated with recurrent episodes of fever that are typically accompanied by pain in the abdomen, chest or joints. This disease most often occurs in people of Mediterranean and Middle Eastern origin, and the first episodes typically begin in childhood [3]. Several studies showed an increase in the frequency of MEFV mutations among children with vasculitic and rheumatic diseases, like inflammatory bowel disease, polyarteritis nodosa, Henoch-Schonlein purpura and juvenile idiopathic arthritis. The MEFV gene has been suggested to play an important role in the pathogenesis of this association [4,5]. To our knowledge, association between SEA syndrome and FMF has not been reported previously. We reported here a rare case of coexisting seronegative enthesopathy and arthropathy syndrome, and familial mediterranean fever.
Case Report
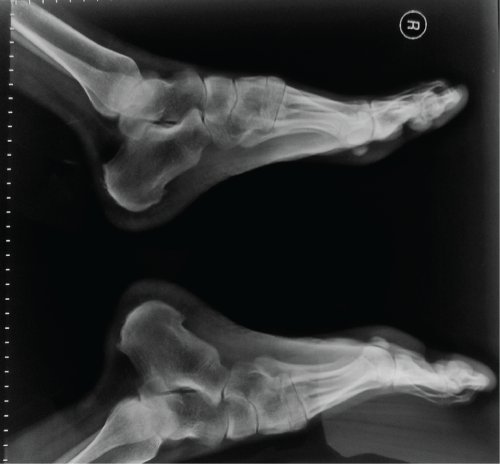
A 20 year old male patient presented, in June 2011, with a history of inflammatory back pain and swelling in his knees and ankles. He reported a 5 years history of low back pain and swelling, and pain on his ankles, wrists and left knee. He had a 1 hours of morning stifness at the begining of his complaints, as well as bilateral alternating buttock pain and heel pain. He did not give a family history of spondyloarthropathies. On laboratory examination, negative values of antinuclear antibodies including ANA and Anti-dsDNA as well as negative values of anti-cyclic citrullinated peptide, rheumatoid factor and HLA-B27 were observed. Routine laboratory examinations including biochemical analyses, urine analyses, complete blood count, eritrocyte sedimentation rate, thyroid functions were in normal limits. Foot radiograms revealed enthesopaties on his heels bilaterally (Figure 1). Magnetic resonance (MR) imagings of his sacroiliac joints did not reveal sacroiliitis. On the other hand, on his MR images of ankles, there were increased effusion surrounding flexor hallucis longus tendon on left side and increased effusion within the subtalar joint space on right side. As a result, SEA syndrome has been diagnosed and he has been followed up for 3 years with a drug treatment including sulphasalazine 3 × 500 mg/day and indomethacin 3 × 25 mg/day. During the follow-up period, in March 2014, he described complaints of recurrent fever and abdominal pain attacks. MEFV gene analysis was performed to the patient and the analysis revealed the existence of homozygous gene mutation of M694V. As a result of clinical and laboratory examinations, coexisting SEA syndrome and FMF was diagnosed and colchicine 0.5 mg twice a day was added his current treatment. After two mounths of treatment, he was painless and he had not complaints of fever attacks. After 6 months follow up period, tarsitis and enthesopathies were persisted.
Discussion
Peripheral enthesitis and arthritis are the major clinical features in the juvenile-onset spondyloarthropathies (SpA). Arthritis often affects the lower extremities and it is often in the form of asymmetric monoarthritis or oligoarthritis. 50-75% of the patients with SEA syndrome progress to juvenile-onset ankylosing spondylitis by 5-11 years after initial presentation [1,6]. The presence of enthesoathy and tarsal disease, in children who have lower extremity arthritis, may differentiate the patients with early-onset spondyloarthritis from JIA patients within the first year of symptoms [7]. In patients with juvenile SpA, tarsal affectation is an episode that is frequently accompanied by acute phase inflammation and, sometimes, by chronic phase ankylosis. The combination of inflammatory and structural tarsal alterations is known as ankylosing tarsitis [8]. In our patient, presence of enthesopathy, arthropathy and inflammatory back pain as well as male gender and juvenile-onset of the disease were the findings compatible with the diagnosis of SEA syndrome [2]. Additionally, foot MR imagings of the patients allerted us on the possibility of a developing ankylosing tarsitis. After three years of follow-up, he developed recurrent fever and abdominal pain attacks and symptoms were resolved with colchicine treatment. Gene analysis of the patient revealed the existence of homozygous gene mutation of M694V and he also meets the diagnostic criteria for FMF [9]. At first glance, these two diseases seemed to be developing separate entities at different times in the same patient. But, we met several studies examining the existence of enthesitis in patients with FMF in pubmed search. In a study, Eshed et al. found that exertional leg pain is a common manifestation of FMF and is frequently associated with an underlying ankle enthesopathy. In their study group, inflammatory markers were significantly higher and M694V homozygosity was more prevalent. Signs compatible with enthesopathy on MRI were significantly more common among the study group (73.5%) and definite SpA was diagnosed in 42.2% of the patients in the study group [10]. Tufan et al. investigated the possible association between FMF and enthesopathy using a sonographic method. Heel was the most common region affected in all patient groups. FMF patients harboring M694V variant had higher ultrasound scores compared to patients with other variants. There was no statistically significant difference in the mean ultrasound scores between healthy subjects and those FMF patients with genetic variants other than M694. They concluded that enthesopathy may not be a feature of general FMF population; rather, it might be specifically associated with the presence of M694V variant [11]. In another study by Ozkan et al. the frequency of enthesopathy in FMF was examined by using an other sonographic method. Their study showed existence of significant enthesopathy in patients with FMF also. They concluded that, the findings support the hypothesis that FMF and SpA may have common inflammatory mechanisms [12]. In the light of the findings of these studies, it seems that there may be common inflammatory mechanisms between FMF and SpA, and M694V mutation may be responsible for this coexistence. Our patient had enthesopathies on his heels. He also had homozygous M694V mutation. So, we think that the existence of these two separate entities in our patient is a coexistence. All studies examining the existence of enthesitis in patients with FMF were performed on patients with FMF in older age group and they investigated the presence of enthesopathy in later stages of the disease. To our knowledge, there are not any studies in the literature evaluating the existence of enthesopathy in the pediatric FMF patients. Future studies will reveal the exact role of MEFV mutations among children with vasculitic and rheumatic diseases.
References
-
Gensler L, Davis JC Jr (2006) Recognition and treatment of juvenile-onset spondyloarthritis. Curr Opin Rheumatol 18: 507-511.
-
Rosenberg AM, Petty RE (1982) A syndrome of seronegative enthesopathy and arthropathy in children. Arthritis Rheum 25: 1041-1047.
-
Hashkes P (2015) Familial Mediterranean Fever 2013 American College of Rheumatology.
-
Salah S, Rizk S, Lotfy HM, El Houchi S, Marzouk H, et al. (2014) MEFV gene mutations in Egyptian children with Henoch-Schonlein purpura. Pediatr Rheumatol Online J 12: 41.
-
Ozen S, Bakkaloglu A, Yilmaz E, Duzova A, Balci B, et al. (2003) Mutations in the gene for familial Mediterranean fever: do they predispose to inflammation? J Rheumatol 30: 2014-2018.
-
Weisman M, Reveille M, van der Heijde D (2006) Ankylosing spondylitis and the spondyloarthropathies. In: Burgos-Vargas R. A companion to rheumatology (3rd edition). Mosby: St Louis pp. 94-106.
-
Burgos-Vargas R, Vazquez-Mellado J (1995) The early clinical recognition of juvenile-onset ankylosing spondylitis and its differentiation from juvenile rheumatoid arthritis. Arthritis Rheum 38: 835-844.
-
Ramirez Gonzalez R, Soto Abraham MV, Ugalde Viteli A, Pacheco Tena C, Burgos-Vargas R (2006) Identification of the best sites for the histopathological study of tarsitis in patients with spondyloarthropathies. Reumatol Clin 2: 164-167.
-
Livneh A, Langevitz P, Zemer D, Zaks N, Kees S, et al. (1997) Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum 40: 1879-1885.
-
Eshed I, Rosman Y, Livneh A, Kedem R, Langevitz P, et al. (2014) Exertional leg pain in Familial Mediterranean Fever: A manifestation of an underlying enthesopathy and a marker of a more severe disease. Arthritis Rheumatol 66: 3221-3226.
-
Tufan A, Mercan R, Tezcan ME, Kaya A, Bitik B, et al. (2013) Enthesopathy in patients with familial Mediterranean fever: increased prevalence in M694 V variant. Rheumatol Int 33: 1933-1937.
-
Ozkan F, Cetin GY, Inci MF, Bakan B, Yuksel M, et al. (2013) Increased enthesopathy in patients with familial Mediterranean fever: evaluation with a new sonographic enthesitis index. J Ultrasound Med 32: 325-332.
