As data on the efficacy of biologics in patients with mild/moderate rheumatoid arthritis are limited, this study was performed to assess the efficacy and safety of tocilizumab plus methotrexate versus tocilizumab monotherapy on disease activity.
Seventy-seven patients with mild/moderate rheumatoid arthritis and an inadequate response (Disease Activity Score 28 > 3.2) to methotrexate were initially enrolled (mean Disease Activity Score 28 3.91 +/- 0.54) and received three infusions of tocilizumab 8 mg/kg every 4 weeks plus methotrexate. Subjects achieving a good/moderate European League Against Rheumatism response after three months of open-label treatment were randomised to Group A (tocilizumab plus methotrexate) or Group B (tocilizumab plus placebo methotrexate). The primary endpoint was the Disease Activity Score 28 change from week 12 to 24. The secondary endpoints included the proportion of patients achieving remission according to the Disease Activity Score 28 and various disease activity indices at week 24.
Sixty-five patients were included in the blinded trial phase. At week 12, the mean Disease Activity Score 28 was 1.51 in Group A (n = 32) and 1.72 in Group B (n = 33). The Disease Activity Score 28 difference between the groups was not statistically significant (p = 0.19). No substantial differences were seen with regard to the secondary endpoints.
Additional tocilizumab treatment led to improvement in patients with mild/moderate rheumatoid arthritis. The study results give no indication that the combination of Tocilizumab with Methotrexate induces a better outcome (preserving the level of disease activity achieved at week 12) in comparison with Tocilizumab monotherapy in patients corresponding to those included into the study.
Mild to moderate rheumatoid arthritis, Inadequate methotrexate response, Tocilizumab
AE: Adverse Events; ANCOVA: Analysis of Covariance; CDAI: Clinical Disease Activity Index; CERTAIN: Certolizumab Pegol in the Treatment of RA: Remission Induction and Maintenance in Patients with LDA; CI: Confidence Interval; CZP: Certolizumab; DAS28: Disease Activity Score 28; DMARD: Disease-Modifying Anti-Rheumatic Drug; ETA: Etanercept; EULAR: European League Against Rheumatism; HAQ-DI: Health Assessment Questionnaire Disability Index; IL-6: Interleukin-6; ITT: Intention-To-Treat; LOCF: Last Observation Carried Forward; MTX: Methotrexate; PP: Per-Protocol; PRO: Patient-Reported Outcome; RA: Rheumatoid Arthritis; RADAI-5: Rheumatoid Arthritis Disease Activity Index-5; SA: Safety Analysis; SDAI: Simplified Disease Activity Index; SF-12v1: 12-Item Short Form Health Survey; TCZ: Tocilizumab; TNF-α: Tumour Necrosis Factor Alpha; TSQM: Treatment Satisfaction Questionnaire for Medication; VAS: Visual Analogue Scale
Over the last decades, there have been major advances in the treatment of rheumatoid arthritis (RA) [1]. While early diagnosis and treatment of the disease have added to improved outcomes, biologic agents also provide improved efficacy in an additional number of patients compared to traditional treatments [2]. However, despite these advances, approximately 30 to 40% of patients fail to achieve an acceptable clinical response or to tolerate the new agents [3].
Disease-modifying anti-rheumatic drugs (DMARDs) – the cornerstone of RA treatment throughout all stages of the disease – maintain or improve physical function and retard radiographic joint damage. For many subjects, however, treatment remains limited by toxicity and/or ineffectiveness [4,5]. During the last fifteen years, biologic compounds that target tumour necrosis factor alpha (TNF-α), B-cells or T-cells have been used successfully to treat RA. Still, a considerable percentage of patients fail to respond to these therapies [6,7].
Overall, there is an obvious medical need for more effective treatments for RA based on a precise understanding of the underlying pathophysiology of the course of the disease. A different therapeutic approach, namely targeting interleukin-6 (IL-6), constitutes the application of tocilizumab (TCZ) - an anti-IL-6 receptor antibody – which has been approved for the treatment of moderate to severe RA [8]. Inhibiting the entire receptor complex prevents IL-6 signal transduction to inflammatory mediators that summon B and T cells. Tocilizumab has a nonlinear pharmacokinetic profile [9].
TCZ has been shown to be more efficacious than methotrexate (MTX) alone in some clinical trials and therefore may offer an alternative, applied as monotherapy, for patients who have experienced intolerability to MTX as well as inadequate clinical response to biologic or non-biologic DMARDs [10]. It has been demonstrated to reduce the rate of progression of joint damage as measured by X-ray and to improve physical function when given in combination with MTX. Clinical efficacy and safety studies with TCZ have been conducted or are ongoing in various disease areas, including adult-onset RA, systemic-onset juvenile idiopathic arthritis, polyarticular juvenile idiopathic arthritis, giant cell arteriitis [11,12].
While the efficacy and safety of biologic DMARDs in general, and TCZ in combination with DMARDs in particular, has been established in moderate to severe RA, only sporadic data are available in patients with mild to moderate RA for some biologic DMARDs [13,14]. Very recently results of a meta-analysis revealed that TCZ as compared to Tumor Necrosis Factor-inhibitors, may be associated with a reduced risk of major cardiovascular events (MACEs), whereas csDMARDs like MTX may be associated with an increased risk of MACEs and particularly stroke. This finding may be related to TCZ's regulatory capability with respect to serum levels of chemerin and adiponectin in RA patients, independently of the disease treatment response [15,16]. Against this background, the present OPTIMISE trial was intended to recruit subjects with mild to moderate RA to study the efficacy and safety of TCZ in patients with less severe disease. The primary objective of this study was thus to assess the efficacy and safety of TCZ in combination with MTX versus TCZ monotherapy to preserve therapeutic response in such subjects and to determine the contribution of MTX by comparing TCZ plus MTX versus TCZ alone in patients who had previously been treated with combined TCZ plus MTX.
The OPTIMISE study was conducted as a local, open-label, phase IIIb trial followed by a randomised, double-blind, placebo-controlled, parallel-group study. The study was conducted according to the guidelines of the Declaration of Helsinki and Good Clinical Practice. The investigators were trained according to the Sponsor's applicable standard operating procedures. The study protocol was approved by the Ethics Committee of Lower Austria (GS4-EK-14/025-2011), and all patients provided written informed consent prior to any study procedures.
One hundred and twelve patients (80 female/29 male/3 missing data), ≥ 18 years of age, ≤ 150 kg of body weight, with mild to moderate active RA of more than one year duration (or radiologic evidence of RA if diagnosis of RA < 1 year) who were currently experiencing an inadequate response (Disease Activity Score 28 [DAS28] ≤ 4.5 and > 2.6) to a stable dose of MTX therapy (15-25 mg/wk) were screened for eligibility. Thirty-five were excluded prior to treatment, the majority of them for screening failures (n = 26). Subsequently, 77 patients, including 12 who were not randomised, received three infusions of TCZ (8 mg/kg) IV every four weeks plus background MTX at the accustomed dose. Table 1 shows the baseline data for these patients.
Table 1: Baseline data (Safety Analysis population; n = 77). View Table 1
Patients having achieved at least a moderate European League Against Rheumatism (EULAR) response [13] after three TCZ infusions were enrolled into the blinded study phase. These 65 subjects (84.4%; 51 female/14 male; mean age 57.5 +/- 11.3 years) were stratified according to age (< 40 years, 40-65 years, > 65 years) and gender in addition to the achievement of good or moderate EULAR response Figure 1.
 Figure 1: Primary disposition of patients (open-label phase).
Figure 1: Primary disposition of patients (open-label phase).
EULAR: European League Against Rheumatism; MTX: Methotrexate; TCZ: Tocilizumab.View Figure 1
Patients randomized to Group A (n = 32) received TCZ 8 mg/kg IV every four weeks plus MTX (15 to 25 mg weekly as in the starting phase), whereas patients randomized to Group B (n = 33) received TCZ 8 mg/kg IV every four weeks plus placebo MTX-Tablets. In order to minimise potential MTX toxicity, all subjects received at least 5 mg oral folic acid weekly for the entire duration of the treatment period. Stable non-steroidal anti-inflammatory drug and oral corticosteroid (≤ 10 mg/d prednisone or equivalent) doses were continued during the trial.
Patients who failed to achieve a good or moderate EULAR response after three months of treatment were excluded from the study and treated according to the investigators' discretion.
The first patient was enrolled on January 17, 2012, and the last patient/last visit was performed on February 13, 2014.
The overall goal of the trial was to assess the effect on disease activity of TCZ plus MTX versus TCZ monotherapy in patients with mild to moderate RA. Therefore, the change in the DAS28 score including the erythrocyte sedimentation rate [17] from week 12 (time of randomisation) to week 24 was chosen as the primary endpoint (Figure 2).
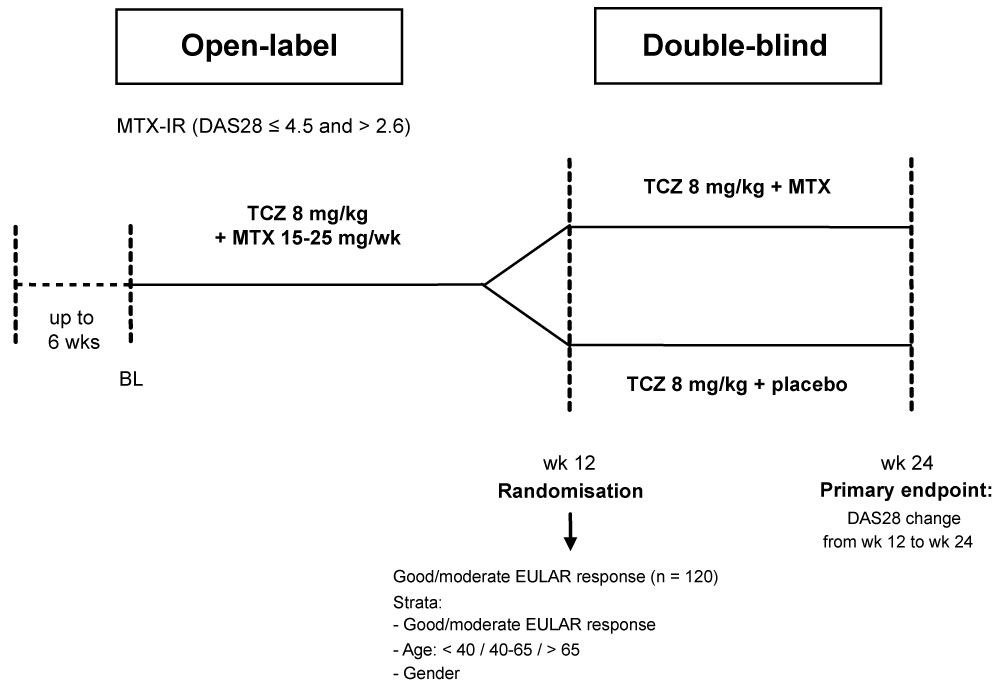 Figure 2: Study design.
Figure 2: Study design.
BL: Baseline; DAS28: Disease Activity Score 28; EULAR: European League Against Rheumatism; MTX: Methotrexate; TCZ: Tocilizumab.View Figure 2
The following patient-reported outcomes (PROs) were recorded: Patients' global assessment of disease activity (VAS), HAQ-DI [18], SF-12v1 [19], RADAI-5 [20], VAS fatigue, VAS pain, and TSQM [21]. The patient global assessment of disease activity contributed to the efficacy endpoints DAS28, SDAI and CDAI [14,22]. The VAS fatigue, HAQ-DI and SF-12v1 were used to assess any improvements in physical and mental health. The TSQM was used to assess the patients' satisfaction with their current treatment. Paper-based PRO questionnaires were administered to the subjects and the resulting PRO data were entered on the electronic case report forms by data management.
The secondary objectives were the following:
• Proportion of patients who achieved DAS28 remission [14] at 24 weeks (DAS28 < 2.6)
• Proportion of patients who achieved Clinical Disease Activity Index (CDAI) remission [22] (CDAI < 2.8) at 24 weeks
• Proportion of patients who achieved Simplified Disease Activity Index (SDAI) remission [22] (SDAI < 3.3) at 24 weeks
• Proportion of patients who achieved Rheumatoid Arthritis Disease Activity Index-5 (RADAI-5) remission [20] (RADAI-5 score ranging from 0 to 1.4) at 24 weeks
• Improvement in physical and mental health according to the Health Assessment Questionnaire Disability Index (HAQ-DI) [18], the 12-Item Short Form Health Survey (SF-12v1) [19], and the Visual Analogue Scales for fatigue (VAS fatigue) and pain (VAS pain)
• Incidence of adverse events (AEs) and serious AEs during the study period
• Patients' satisfaction with treatments according to the Treatment Satisfaction Questionnaire for Medication (TSQM) [21]
For safety reasons, laboratory parameters were determined every four weeks, the normal values were applied according to Austrian Society of Quality Assurance norms. All laboratory analyses were performed by the local laboratories at the study sites, except those of matrix metalloproteinase-3 which were performed at the Medical University of Graz.
Three populations were statistically analysed:
• Safety analysis (SA) population: Patients who had been given an ID and at least one dose of study medication in the open-label phase;
• Intention-to-treat (ITT) population: Patients randomised to either treatment arm if at least one dose of study medication was administered after randomisation;
• Per-protocol (PP) population: The subset of ITT patients who completed the study without any major protocol violations.
Missing DAS28 values after randomisation were replaced by applying the last observation carried forward (LOCF) principle. Other types of missing data were not imputed.
The randomised study had a confirmatory status (two parallel groups: Group A = TCZ plus MTX vs. Group B = TCZ plus placebo). The primary efficacy analysis tested the null hypothesis that there is no difference in DAS28 change from week 12 to week 24 between the two groups against the alternative that there is a difference at the 5% significance level.
A sample size estimation assuming a difference in means of 0.5 DAS28 change from week 12 to week 24 between Group A and Group B and a common standard deviation of 0.95 resulted in a need for 60 patients in each group (type I error = 5% two-sided, type II error = 19%). Considering a 20% drop-out rate before randomisation (especially by not achieving a good or moderate EULAR response at week 12), 150 patients were to be enrolled.
All data sets of continuous variables were checked for normal distribution (Kolmogorov-Smirnov test with Lilliefors significance correction, type I error = 5%) and for variance homogeneity (Levene test, type I error = 5%). For group comparisons of continuous variables, the t-test was used if normality and variance homogeneity could be assumed. Welch's t-test was used if normality and no variance homogeneity could be assumed. Otherwise, the exact Mann-Whitney U test was used. Categorical variables were compared with Fisher's exact test or the exact chi-square test.
According to the nature of the data sets, group comparison of the primary endpoint (DAS28 change from week 12 to week 24) was performed with the t-test. Due to a substantial difference in the DAS28 at week 12, a parametric analysis of covariance (ANCOVA) with the baseline DAS28 as covariate complemented the statistical approach.
The type I error was not adjusted for multiple testing. Therefore, all results of inferential statistics are descriptive only, except for hypothesis testing for the primary endpoint.
Statistical analysis was performed using the open-source R statistical software package, version 3.1.1 (The R Foundation for Statistical Computing, Vienna, Austria).
A total of 62 of the 65 patients who had been included in the double-blind phase of the trial (the ITT population) finally completed the study.
There was a total of 31 protocol violations after randomisation - the most frequently being violations of inclusion/exclusion criteria and including three dropouts - which led to an exclusion of 22 subjects from the ITT population.
No substantial differences were identified between the groups in the ITT population with respect to their demographic data (age at screening, gender). At baseline (time of randomisation), substantial differences were seen in TSQM side-effects and TSQM convenience (see Table 1), which were obviously due to premedication among the inadequate responders to MTX.
The ITT population was used for the primary efficacy analysis (ITT analysis). Sixty-five patients achieved a moderate (n = 6) or good EULAR response (n = 59) at week 12 (ITT population). In these patients, the mean DAS28 at week 12, the time of randomisation, amounted to 1.51 in Group A (n = 32) and 1.72 in Group B (n = 33), respectively. The numerical change in the DAS28 score from week 12 to week 24 was not statistically significant, but slightly positive in Group A and slightly negative in Group B, respectively (0.17 ± 0.83 vs. -0.16 ± 1.13; 95% confidence interval [CI] for the difference: -0.16 - 0.82). The null hypothesis that no difference in DAS28 change can be seen from week 12 to week 24 between the two treatment groups was not falsified (p = 0.19; ANCOVA: p = 0.30).
In addition, the analysis of the PP population provided very similar results (0.12 ± 0.87 vs. -0.19 ± 0.97; 95% CI for the difference, -0.26 - 0.89; p = 0.28; ANCOVA: p = 0.33).
As to the secondary endpoints – i.e. the proportion of patients in remission, improvement of function, mental health and satisfaction with treatment – no pronounced differences were seen between the MTX- or placebo-treated patients. In general, most of the secondary endpoints showed a very slight tendency towards better results in Group B (the placebo group). No new signals were detected regarding tolerability (see Table 2 and Figure 3).
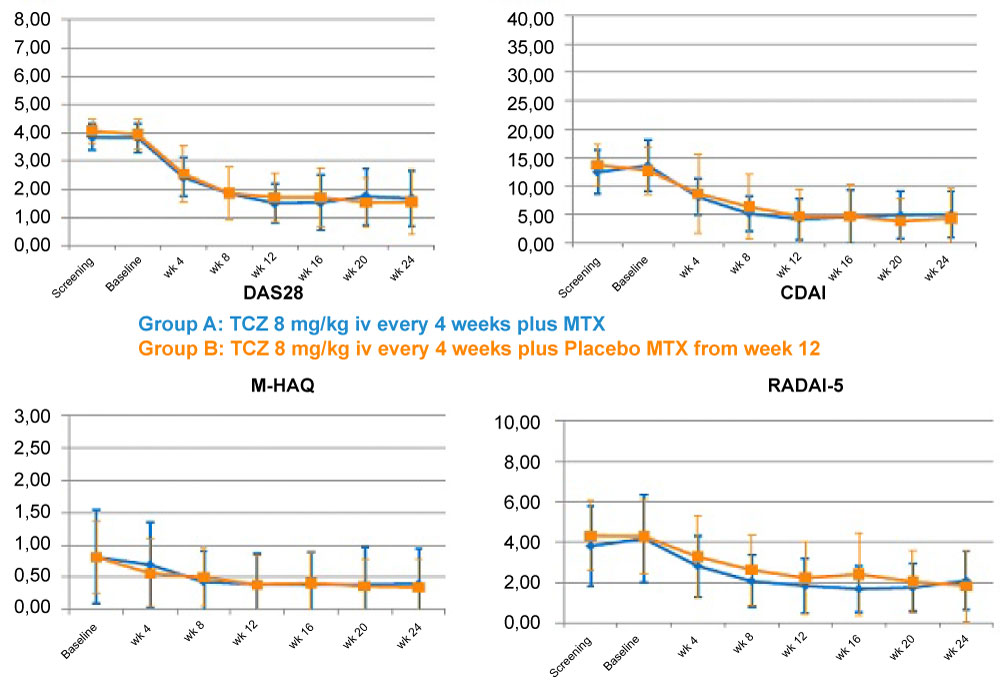 Figure 3: Course of the RA indexes and PROs during the study.
Figure 3: Course of the RA indexes and PROs during the study.
CDAI: Clinical Disease Activity Index; DAS28: Disease Activity Score 28; M-HAQ: Modified Health Assessment Questionnaire; MTX: Methotrexate; PRO: Patient-reported outcome; RA: Rheumatoid arthritis; RADAI-5: Rheumatoid Arthritis Disease Activity Index 5; TCZ: Tocilizumab.View Figure 3
Table 2: Secondary endpoints (ITT population, n = 65). View Table 2
Seven serious AEs were reported in the SA population (Group A: 2, Group B: 5), which comprises all patients having received at least a single dose of TCZ (n = 77).
Substantial differences in common AEs were seen only with respect to "blood and lymphatic system disorders" (Group A: 14, Group B: 6; p = 0.033) and "investigations" (Group A: 14, Group B: 24; p = 0.024). The other most frequent AEs were 44 "infections and infestations" (Group A: 20, Group B: 22, not randomised: 2) and 20 "gastrointestinal disorders" (Group A: 6, Group B: 12, not randomised: 2).
No death occurred during the entire study. In either treatment group, one serious AE with a possible relationship to one of the investigational drugs was reported: one possible relationship with MTX for an alanine transaminase elevation in Group A and one possible relationship with both compounds (TCZ, MTX) for an urinary tract infection in Group B, which was the only serious AE leading to study termination in this particular patient (see Table 3).
Table 3: AEs in treatment groups during the blinded phase of treatment. View Table 3
A plethora of clinical studies and also meta-analyses has addressed the safety and efficacy of biologic DMARDs of any type in patients with highly active RA [7]. In fact, their benefits for patients are beyond any doubt. Although persistently moderately active RA is known to not constitute a benign disease [23], only sporadic reports have dealt with RA patients in the low or moderate disease activity range [11,12,24]. It is well known that lower disease activity, negative rheumatoid factor, shorter disease duration, younger age, etc., constitute predictors of advantageous treatment outcome, the disease activity level being amongst the strongest. Yet if the only reasons not to apply the unquestionably costly biologic compounds to possibly less affected patients are economic, is it then justified to deprive such patients of this type of treatment? One prerequisite to reply to such issues is to affirm each drug's efficacy and safety.
Regarding TCZ, in particular, another question can be regarded as topical as the previous one. TCZ monotherapy has been shown to be more effective than MTX alone; therefore, it is interesting to explore whether co-medication with MTX is necessary to maintain favourable treatment response [24]. Considering this background, the OPTIMISE trial should redound to a more clearly evidence-based application of TCZ.
This trial was performed as an Austrian country-wide study including 14 centres and reflecting daily clinical routine. In fact, a higher degree of homogeneity with respect to patients and their social circumstances can be assumed in comparison with two international multicentre trials: the PRESERVE study investigating Etanercept (ETA) and the Certolizumab Pegol in the Treatment of RA: Remission Induction and Maintenance in Patients with LDA (CERTAIN) trial investigating Certolizumab (CZP) in the respective RA subgroups [12,13]. Comparing the baseline date of the three trials, the subjects in OPTIMISE were older and less severely diseased, as expressed by the DAS28 and HAQ-DI values [12,13], while the gender distribution appears to be similar, indicating that the patients in the OPTIMISE trial more closely matched those originally intended to be studied.
As with the two TNF-α inhibitors applied in the PRESERVE and CERTAIN studies, a high number of patients responded to TCZ treatment within 12 weeks: A EULAR response was recorded in 84.4% overall, of whom 91% achieved a good response. Although this part of the trial was performed in an open manner, the high percentage of EULAR responses gives a robust indication of the efficacy of TCZ as an additional medication to primarily ineffective MTX in RA patients with moderate or low disease activity. As expressed by the composite indexes, the week-24 remission rates in the OPTIMISE trial were considerably higher than with ETA or CZP [12,13] and, interestingly, numerically higher in the TCZ monotherapy group. Additionally, the relevance of this improvement is underlined by the fact that the courses of composite indices and PROs parallel each other to a very high degree, indicating that the benefit is not only arithmetic, but that the subjects indeed experienced that benefit. Also, the percentage of patients in remission as expressed by the RADAI-5 was in the range of the CDAI remission. This can be considered remarkable with TCZ, as it relativizes the discussion about the advantage IL-6 blockers may have if acute phase reactants are components of indices.
It had been anticipated that TCZ treatment would lead to significant improvements if added to ineffective MTX [24]. However, the question whether the combination of TCZ with MTX would perform better than TCZ monotherapy in maintaining treatment response seemed to be of even greater interest. At any rate, our study design – TCZ plus MTX in the open-label phase followed by MTX cessation - served to rule out placebo effects, as only responders were finally included in the blinded phase of the investigation.
No substantial differences were seen when comparing the DAS28 and other index values at week 12 and week 24, no matter whether MTX was co-administered with TCZ or placebo. Not only disease activity in a stricter sense, but also quality of life, as expressed by the SF-12, and satisfaction with treatment was shown to be similar, and independent of MTX co-medication. These findings can be seen in line with previous results indicating a favourable efficacy of TCZ over MTX, and no additional benefit of the combination in patients with RA, which seems to be a unique feature of IL-6 blockers, and TCZ in particular, compared to other biologic DMARDs, such as TNF blockers and co-stimulation blockers. Finally, the achievement of the primary study objective offers another perspective for the applicability of TCZ monotherapy in RA patients.
No new signals were detected with respect to safety, which can be regarded of particular importance in a less severely diseased patient population. Fortunately, no death and only seven serious AEs occurred during the course of the trial. Three of them, namely elevation of liver enzymes, urinary tract infection and stroke, were possibly attributed to the study drugs. The majority of AEs were mild infections and infestations, as expected, blood pattern changes and gastrointestinal disturbances, none of which resulted in a withdrawal of the study drugs.
Our data showed a slight tendency towards a better outcome for the patients in the group receiving TCZ alone without MTX. Although the final sample size in the study was considerably lower than initially planned in the protocol (65 actually enrolled vs. 150 planned patients), it is very unlikely that superiority of TCZ plus MTX would have been shown with the full sample size of 150. Considering the current 95% CI of the DAS28 group difference between week 12 and 24 of -0.16 - 0.82 favouring TCZ and placebo, the statistical likelihood of a clinically meaningful DAS28 difference of > 0.6 according to the EULAR recommendations in favour of the TCZ plus MTX regimen can be considered far below 5%.
The non-inferiority (however not statistically proven) of the TCZ regimen without MTX is also supported by the findings in the secondary endpoints, as they show results in line with those of the primary endpoints.
In summary, this two-phase trial consisting of an open-label and a double-blind phase was successful in achieving its objectives. On the one hand, the efficacy of TCZ in RA patients with low or moderate disease activity was demonstrated. On the other hand, the primary objective - the equivalence of TCZ monotherapy and the combination with MTX in maintaining treatment response – could be demonstrated.
Are there practical consequences to be drawn from this study? First, if financial issues are not the primary maxim physicians act on, biologic DMARDs constitute the possibility to optimise the treatment regime in RA patients who fail to achieve a low level of disease activity. Second, TCZ monotherapy was shown to be as efficacious as the TCZ-MTX combination in maintaining beneficial treatment response, which should be considered in the presence of long-term therapy and drug reduction plans, but also if MTX proves intolerable. Third, RA patients at moderate disease activity do not seem to be at a greater risk if TCZ therapy is initiated, indicating a favourable risk-benefit ratio for the application of this treatment regime.
In conclusion, additional TCZ treatment led to improvements in patients with mild to moderate RA and an inadequate response to MTX. TCZ monotherapy was seen not less effective than the combination with MTX to preserve the level of disease activity achieved at week 12. However, the uncertainty of the results is still large enough to allow speculation on even very small positive, though clinically negligible, effects of the combination.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
BFL, RL, PF, MH, OZ, UR, WS and WBG made substantial contributions to study conception and design, as well as to data acquisition, analysis and interpretation. BFL, RL, PF, UR and WS drafted the article, and MH, OZ and WBG revised it critically for important intellectual content. All authors gave approval of the version of the article to be published.
The abstract of this paper was presented at the 2015 ACR/ARHP Annual Meeting in San Francisco as a conference talk with interim findings. The abstract (No. 1039) was published in Arthritis Rheumatol. 2015; 67 (suppl 10).
The study protocol was approved by the Ethics Committee of Lower Austria (GS4-EK-14/025-2011), and all patients provided written informed consent prior to any study procedures.
Not applicable.
BFL has received research grants from Celltrion, Schering-Plough, Wyeth, Roche, MSD, Centocor, Abbott, Amgen, Aesca, as well as honoraria not exceeding EUR 5,000 each from Centocor, Abbott, Amgen, Aesca, UCB, Roche, MSD, Celltrion, GSK Schering-Plough, Wyeth, Pfizer, BMS, Jannssen-Cilag, Eli-Lilly, Novartis, Sandoz, Gebro and Celgene. WBG has neither competing interests nor any personal or financial interest regarding this publication. He has received speaker fees for educational lectures from all major companies in the field not exceeding EUR 10,000 per year. UR is an employee of Roche Austria GmbH. All other authors have not declared any competing interests.
Trial registration: ClinicalTrials.gov identifier: NCT01587989. This work was supported by Roche Austria GmbH, Vienna, Austria.
The authors gratefully acknowledge their co-investigators' as well as their medical writer's invaluable contributions to the OPTIMISE trial: Elke Böttcher (Vienna), Hans-Peter Brezinschek (Graz), Hans Bröll (Vienna), Ludwig Erlacher (Vienna), Peter Knoflach (Wels-Grieskirchen), Wolfgang Kranewitter (Wels-Grieskirchen), Andrea Leeb (Hollabrunn), Herwig Pieringer (Linz), Franz Rainer (Graz-Eggenberg), Martin Reinwein (Hollabrunn), Judith Sautner (Stockerau), Clemens Scheinecker (Vienna), Josef S. Smolen (Vienna), Ulrike Stuby (Linz), Jeannette Wolf (Vienna), Jochen Zwerina (Vienna) and Karl Thomanek (Vienna, medical writer).