Lymphomatoid granulomatosis (LYG) is a rare, Epstein barr virus (EBV) associated, angiocentric B cell proliferation, which usually involves the lung but may also involve, more rarely, the central nervous system. Its malignant potential is uncertain, and seems to be linked to the number of EBV positive B cells.
We report the case of a 32-years-old patient, with an antecedent of LYG grade 2, involving lung, who presented with a brain mass lesion one year later, which appears to be a grade 1 LYG without systemic lesion associated. Treated with dexamethasone, the patient underwent a rapid clinical recovery as well as a complete radiological response, maintained two years after the diagnosis.
Little is known about LYG affecting the CNS, and even less about the best therapeutic strategy for those patients. A review of the literature has been conducted and a summary of the therapeutic options for this extremely rare disease is presented. This case is unusual, regarding the initial imaging presentation, mimicking a primary aggressive brain tumor. It does also emphasize the fact that aggressive treatment could be avoided for some selected patients.
Lymphomatoid granulomatosis, Central nervous system, Epstein-Bar virus, Corticosteroids
Lymphomatoid granulomatosis (LYG) is a rare EBV-driven extranodal lymphoproliferative disease characterized by a polymorphic lymphoid proliferation with infiltration of artery and veins. According to the WHO classification, it is considered as a form of lymphoproliferative disorders under the general heading of B-cell proliferation of uncertain malignant potential. LYG are sub classified in a three scale grading system according to the number of EBV-positive B cells. Grade 1 displays no or rare EBV-positive cells, whereas grade 2 and 3 have an increased number of EBV-positive cells. The course of the disease is uncertain and seems to depend on the grade of the disease. LYG predominantly involves the lung, but can also affect other organs [1-3].
Involvement of the Central nervous system (CNS-LYG) has been described in one third up to 50% of the patients [1,4]. However, little is known about the prognosis and the appropriate management of CNS-LYG.
Here we present a case of a patient that, despite an initial presentation mimicking an aggressive brain tumor, achieved a complete and durable response with corticosteroid treatment alone. We performed a review of CNS-LYG cases published in the English literature within the last 15 years. We included only cases with adequate data regarding histological grading, treatment modalities and follow up, in order to assess current knowledge on therapeutic strategies recommended for those patients.
We entered the following search items into PubMed and Google Scholar search engines: lymphomatoid granulomatosis, central nervous system and cerebral, and limited the search for the period of 2002 to 2017. Reports of adult patients that included sufficient data regarding pathological findings (according to the current classification), treatment modalities and follow up period, served for this review.
A 32-year-old man was referred to our institution in November 2015 because of appearance of several, progressive, neurological symptoms within a period of weeks: headaches, slight left hemiparesis and mild cognitive impairment. Since October 2014, he was followed in another institution for lung lymphoid granulomatosis, grade 2 (lung biopsy displays 18% EBV- positive cells using FISH analysis). FDG PET-CT (whole body scan without study of the brain), performed on 31/01/2015 showed a nodular infiltration of both lungs with an intensive bilateral lung uptake of FDG tracer (Figure 1A and Figure 1B). Due to absence of clinical symptoms he did not received any specific treatment and was followed clinically and radiographically with subsequent spontaneous resolution of lung lesions.
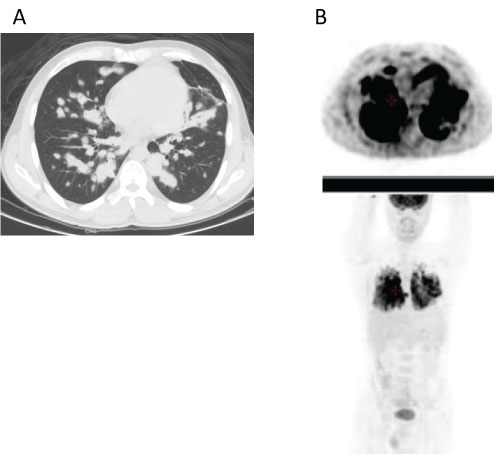 Figure 1: CT scan performed on 31/01/2015 demonstrated nodular infiltration of both lungs (Figure 1A) with a high uptake of FDG tracer on PET imaging (Figure 1B).
View Figure 1
Figure 1: CT scan performed on 31/01/2015 demonstrated nodular infiltration of both lungs (Figure 1A) with a high uptake of FDG tracer on PET imaging (Figure 1B).
View Figure 1
On admission, neurological examination was remarkable for papilledema and slight left hemiparesis as well as mild cognitive impairment (mainly short term memory difficulties). Routine laboratory tests (CBC and biochemistry) were normal. Human Immunodeficiency virus was negative. Brain MRI showed two contrast enhancing lesions containing a thick ring enhancement that affected the right frontal and temporo-parietal lobes. The lesions also involved the corpus callosum and were associated with brain edema and a significant mass effect (Figure 2A and Figure 2B). The diagnosis of a possible primary aggressive brain tumor (multifocal glioblastoma or lymphoma) was initially suggested.
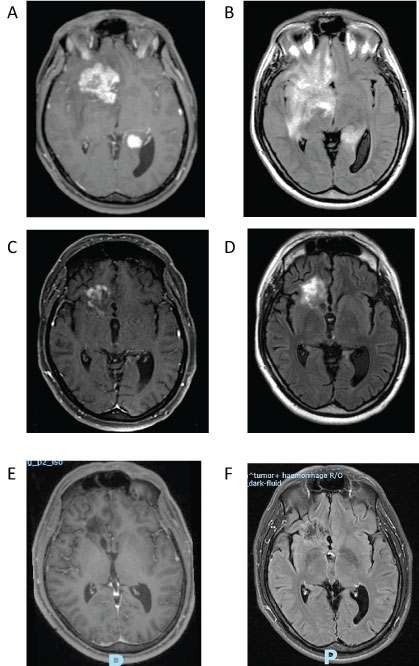 Figure 2: Enhanced T1-weighted MRI sequences at diagnosis (November 2015) demonstrated two enhancing lesions (2A). Associated with a significant edema and mass effect on FLAIR sequence (2B). Highly suggestive of a bifocal aggressive brain tumor.
Figure 2: Enhanced T1-weighted MRI sequences at diagnosis (November 2015) demonstrated two enhancing lesions (2A). Associated with a significant edema and mass effect on FLAIR sequence (2B). Highly suggestive of a bifocal aggressive brain tumor.
Follow up MRI performed three months after steroid therapy (February 2016) demonstrated a marked improvement and reduction of the enhancing lesions on enhanced T1-weighted sequences (2C) and reduction of brain edema and mass effect on FLAIR sequences (2D).
A complete response with disappearance of the enhancing lesions (2E) On enhanced T1-weighted sequences as well as resolution of hyperintensity FLAIR signal (2F) was demonstrated on MRI performed on September 2016.
View Figure 2
A stereotactic biopsy was obtained from the frontal lesion on November 15, 2015, before any steroid treatment. Pathology revealed an infiltration of small and large lymphocytic cells. Immunohistochemistry revealed numerous CD3 positive cells, including scattered small CD20 positive cells and a few CD30 positive large cells. We performed a PCR analysis both for B-cell and T-Cell clonality in order to exclude diagnosis of lymphoma. PCR analysis of IgH gene rearrangement and of T-cell receptor gamma gene rearrangement revealed B-cell, as well as T-cell, polyclonality. Ki67 was estimated as 20%. Comparison between the slides from the brain biopsy and the slides from the lung biopsy (performed one year before) revealed a similar histologic pattern. However in the brain (contrary to the lung biopsy) EBV search was negative: in both staining for EBV as well as in CISH-EBER analysis. The diagnosis was compatible with a grade 1 CNS lymphomatoid granulomatosis due to lack of EBV-positive cells. Since the diagnosis of CNS involvement was based on a biopsy sample, it is not possible to decisively exclude the presence of EBV-positive cells due to possible regional diversity in the amount of EBV- positive cells within the lesion.
Immediately after the biopsy, the patient was treated with dexamethasone (starting dose 12 mg/day) with a rapid recovery of his neurological symptoms and signs.
Whole body PET-FDG CT, as staging of the disease, was only performed on 01/01/2016, 6 weeks later and revealed no systemic involvement, especially no involvement of the lung. This suggest a recurrence involving only CNS. However this imaging, because of the delay and of the concomitant steroid treatment is difficult to analyse.
Brain MRI follow-up showed a progressive improvement. First MRI, performed 3 months later in February 2016, showed a marked improvement (Figure 2C and Figure 2D) and consecutive MRI studies revealed continuous reduction of the enhancing lesion with a complete response documented in September 2016 (Figure 2E and Figure 2F). Steroid treatment was gradually tapered down and discontinues one year after diagnosis. The patient remained in remission at the last follow up in October 2017.
Our patient's initial neurological and imaging presentation was challenging because it mimicked at first a primary brain tumor, with a heterogeneous enhancing brain lesion, associated with a significant mass effect. According to literature (case reports and also a retrospective study conducted by Patsalides et al, including 25 patients with pathologically confirmed LYG), multiple intraparenchymal T2/FLAIR lesions with only punctuate enhancement, or abnormal leptomeningeal enhancement are usually described. However cases of brain masses, as in our case, were also reported [4-6].
Data concerning treatment and follow-up for patients with CNS-LYG is also scares and consists mostly of case reports or small retrospective studies. Our literature review found 25 other cases that fulfilled the inclusion criteria (Table 1) [3-20]. Including our case, there are 16 men and 10 women with a median age at diagnosis of 50.5 years (range: 19-80 years).
Table 1: Literature review: Cases of CNS LYG. View Table 1
It is interesting to note that 16 patients (61%) had a diagnosis of primary CNS-LYG with an isolated CNS involvement at time of diagnosis. This high proportion probably represents a publication bias because those cases are more likely to be published than secondary CNS-LYG that follows systemic disease.
The median follow up was 31.4 months (range: 1 week to 18.4 years) with 22 patients still alive and 4 deceased at time of reporting. Treatment modalities varied greatly: at least 9 patients underwent surgical resection, 10 were treated with chemotherapy (5 of them received also cranial irradiation), another 2 patients were treated with radiotherapy alone, 2 received rituximab monotherapy, 7 steroids as a single modality and one a combination of steroid and interferon α-2b. Three patients were not treated, one with grade 3 LYG, due to a rapid clinical deterioration [20] (died a week after the histological diagnosis), the two others, with grade 1 LYG, had regressed. One of them was treated with anti-retroviral therapy for AIDS, and had a spontaneous regression without other specific therapy [7] and the other one had a complete remission within a few months without any treatment [13]. It is also notable that 2 patients received immunosuppressive therapy after renal transplantation before developing CNS-LYG (case 16 and 21) [16]. They were treated with steroid therapy in addition to the immunosuppressive drugs which lead to a partial response.
Systemic LYG remains a rare disease, with a highly variable clinical course and without a well-established standard of care. Grading is based on the number of EBV-positive cells and has relevant prognostic implications. As such, it is a key factor for selection of the therapeutic strategy. Grade 1 LYG, with no or rare EBV positive cells, is supposed to be an indolent disease and can sometimes undergo a spontaneous regression. Pending on symptomatology it may sometimes be followed without aggressive therapy. On the contrary grade 3 LYG, which contains numerous EBV-positive cells, is usually considered as a variant under the category of diffuse large B-cells lymphoma and therefore requires an intensive therapeutic approach. Therapeutic approaches for grade 2 are less clear as they can also display an indolent behavior [1-3].
Treatment recommendations for CNS-LYG are even less clear. Grading seems to carry similar prognostic implication for CNS-LYG as for systemic disease. Yet, there is no consensus about prognosis and the best therapeutic approach for those patients.
In Table 1 CNS-LYG cases are sorted by the pathology grading. Half of the reported cases (13/26 50%) were of low grade. Prognosis of grade 1 patients seems to be better than grade 2 or 3 judging from the reported follow-up period and time to progression. Unfortunately, survival has not been reported in the majority of cases and therefore it is impossible to get a clear concept on overall survival.
Therapeutic strategies differed between the two groups (grade 1 vs. grade 2 or 3). Only 4 of 13 patients (33%) with grade 1 CNS-LYG received an intensive therapy (chemotherapy or radiotherapy) versus 8 of 13 patients (61%) with grade 2 or 3. Steroids as monotherapy were used in 5 of 13 patients with grade 1 (including our patient) and in 2 of 13 patients with grade 2 or 3. Data on response to treatment is available for only 17 patients. Seven patients displayed a complete response (4 patients with grade 1 and 3 with grade 2 or 3), seven a partial response, and three a progressive disease. Patients with progressive disease had a short survival.
In conclusion LYG is a rare disease and CNS involvement affects about one third of the patients. There is no consensus on standard of care for patients with CNS-LYG. It seems that the grading of LYG helps to select the therapeutic strategy. In grade 1 CNS-LYG, chemotherapy and radiotherapy can probably be deferred and steroid therapy alone may induce a long term remission as was the case in our patient. The possible role of rituximab as an optional monotherapy remains unclear and needs to be explored in the future. On the other hand, high-grade CNS-LYG is probably best managed by combination chemotherapy and immunotherapy but still tends to have inferior outcomes. However, the data remains rare and there is clearly a need to improve our understanding of this rare entity and to compose guidelines for selection of therapeutic strategies for this rare disease.