There are quite a few entities which can present as dura-based masses. Most common being meningioma, others are gliosarcomas, solitary fibrous tumor, leiomyosarcoma, haemangiopericytomas and melanocytomas. Recently, a number of neoplastic and non-neoplastic entities have been reported that radiographically and clinically mimic meningiomas. This is a rare case of glioblastoma with rhabdoid features presented as dura-based tumor. Even though the imaging helps to narrow down the diagnostic possibilities, awareness of various entities is necessary as they have a wide range of prognoses and differing management strategies.
Dura, Meningioma, Glioma, Rhabdoid, Tumor, Imaging
The gliomas presenting as dura-based tumor are quite uncommon. Most of them are parenchymal lesions with invasion of dura [1]. They are most of the time are gliosarcomas which compose about 1.7% to 2.3% of glioblastomas 18% to 20%. Glioblastomas with rhabdoid cell morphology usually mimic rhabdoid meningioma or other tumors with unknown histogenesis. Most of the time imaging gives certain key features which increase the probability of a lesion being a meningioma which include intralesional calcifications, skull hyperostosis, local dural enhancement and increased perfusion. Arriving at right diagnosis helps in the prognosis and management.
A 71-year-old lady presented with a sudden onset of complete loss of power in the left upper and lower limb for an hour. She was rushed to the hospital and imaged for a stroke. Rather than presenting with signs of an infarct or bleed she was seen to have a paramedial dural based lesion compressing on the motor strip, with significant perilesional oedema (Figure 1). The lesion had a focus of bleed which possibly explained the sudden deterioration of symptoms. Perfusion imaging showed no significant increase in Lesional blood flow (Figure 2) and contrast showed a uniform enhancement (except for the region of bleed). No evidence of stroke was present on DWI imaging, although multiple old infarcts consistent with her age, but were seen not corresponding to the present clinical scenario. MR spectroscopy was also done which suggested a neoplasm on Non-neural origin with high cellular necrosis (Figure 3).
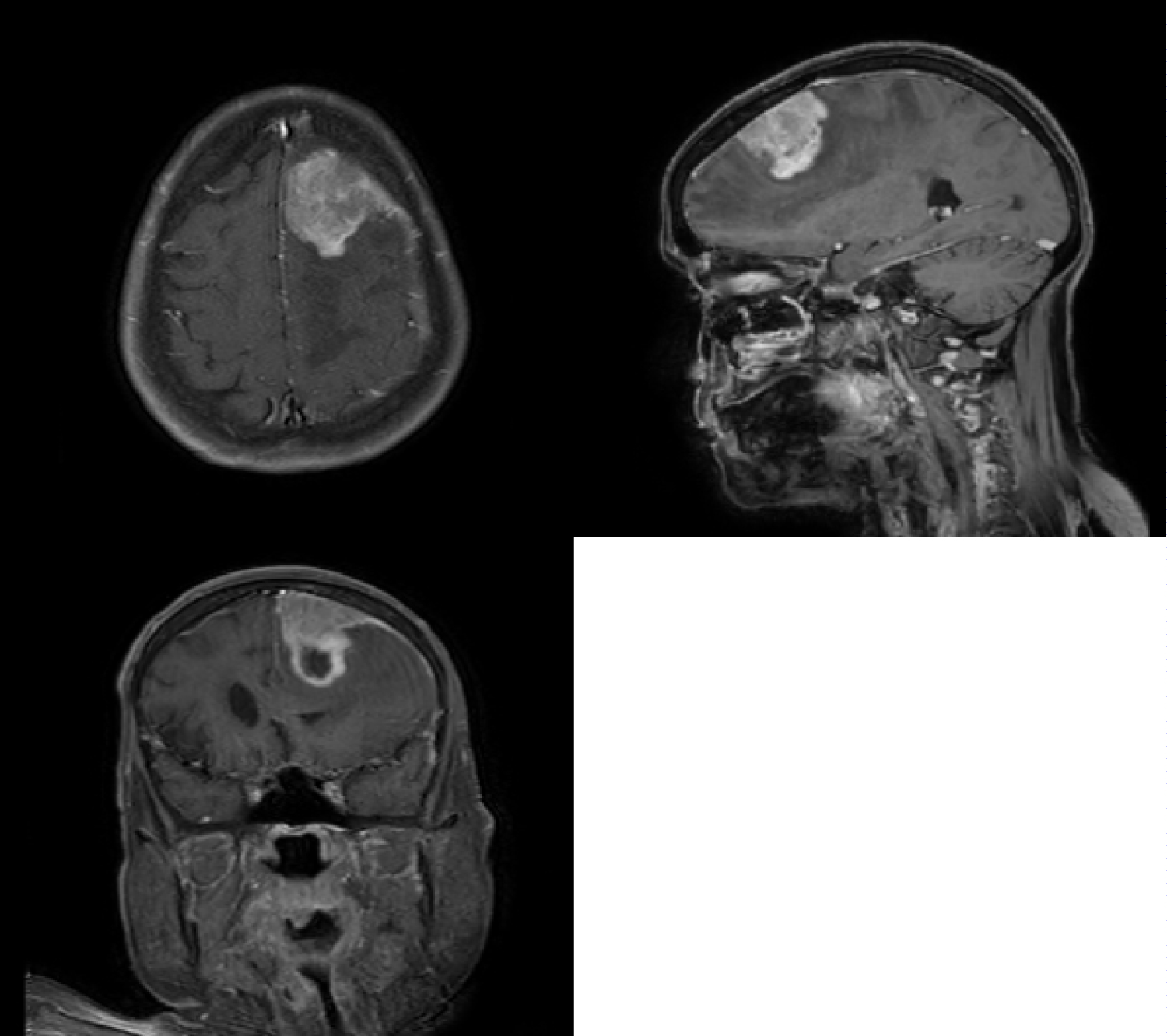 Figure 1: Contrast MRI in Sagittal, Coronal and Axial Planes showing the lesion in the parasagittal region with uniform enhancement and a dural attachment.
View Figure 1
Figure 1: Contrast MRI in Sagittal, Coronal and Axial Planes showing the lesion in the parasagittal region with uniform enhancement and a dural attachment.
View Figure 1
 Figure 2: Perfusion MRI sequences show no significant increase in blood intake by the lesion.
View Figure 2
Figure 2: Perfusion MRI sequences show no significant increase in blood intake by the lesion.
View Figure 2
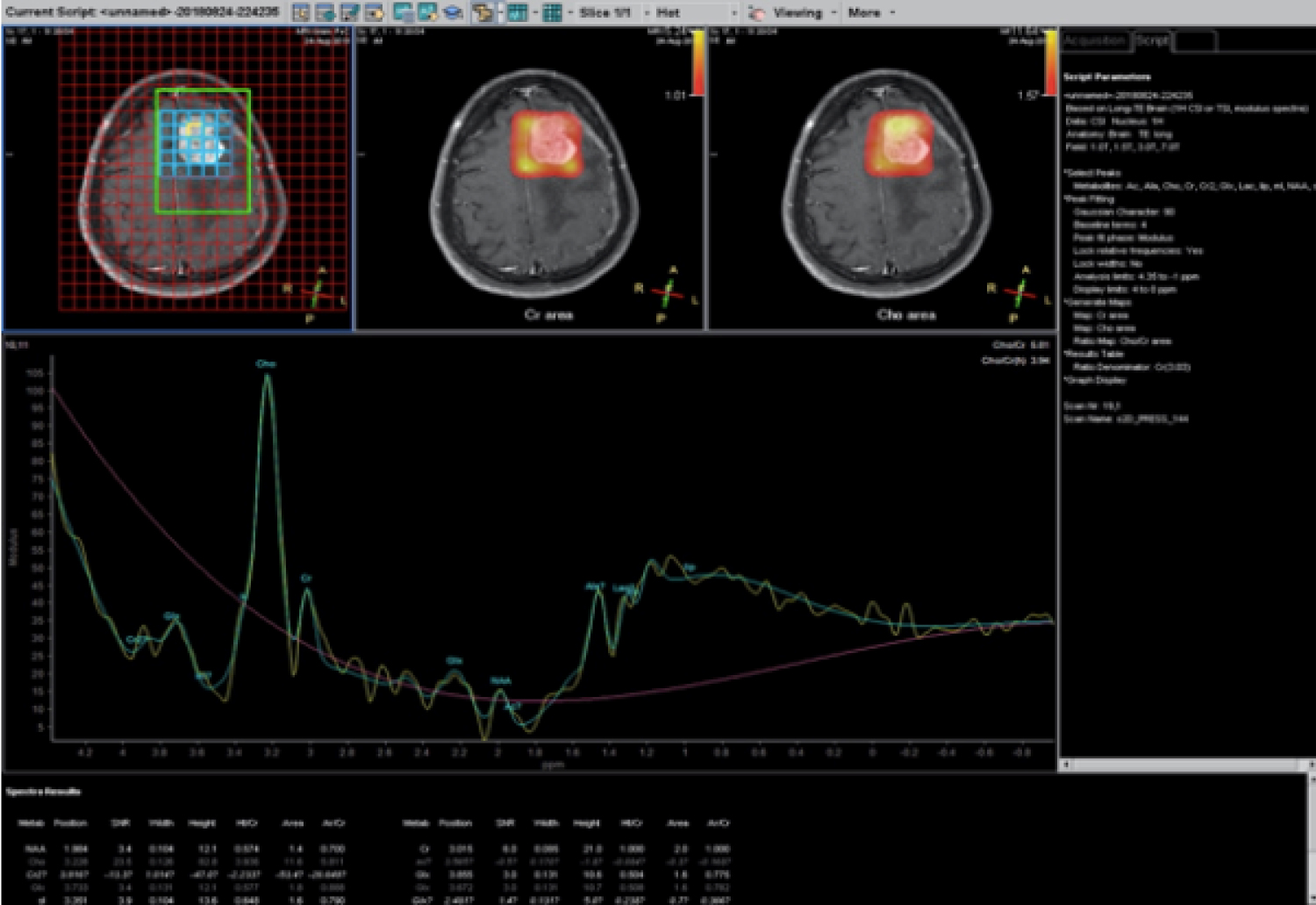 Figure 3: Spectroscopy showing peaks of Lactate and no NAA suggestive of high cell turnover and no neural elements.
View Figure 3
Figure 3: Spectroscopy showing peaks of Lactate and no NAA suggestive of high cell turnover and no neural elements.
View Figure 3
The patient was explained the nature of the illness and counselled for surgery. She underwent a paramedian craniotomy and complete excision of the lesion. There was no dural sinus invasion, and the sinus wall was free (Figure 4). The lesion was moderately vascular with no invasion of the pia or arachnoid. A diagnosis of meningioma was made on table and a Simpson's grade 2 Excision was done with coagulating the dural attachment. Watertight dural closure was done and the patient was extubated on table and shifted to the HDU for recovery. She was discharged in a few days to review in OPD. Home physiotherapy was started aggressively, and she improved well. Her hemiplegia improved to grade 3 power in both upper and lower limbs.
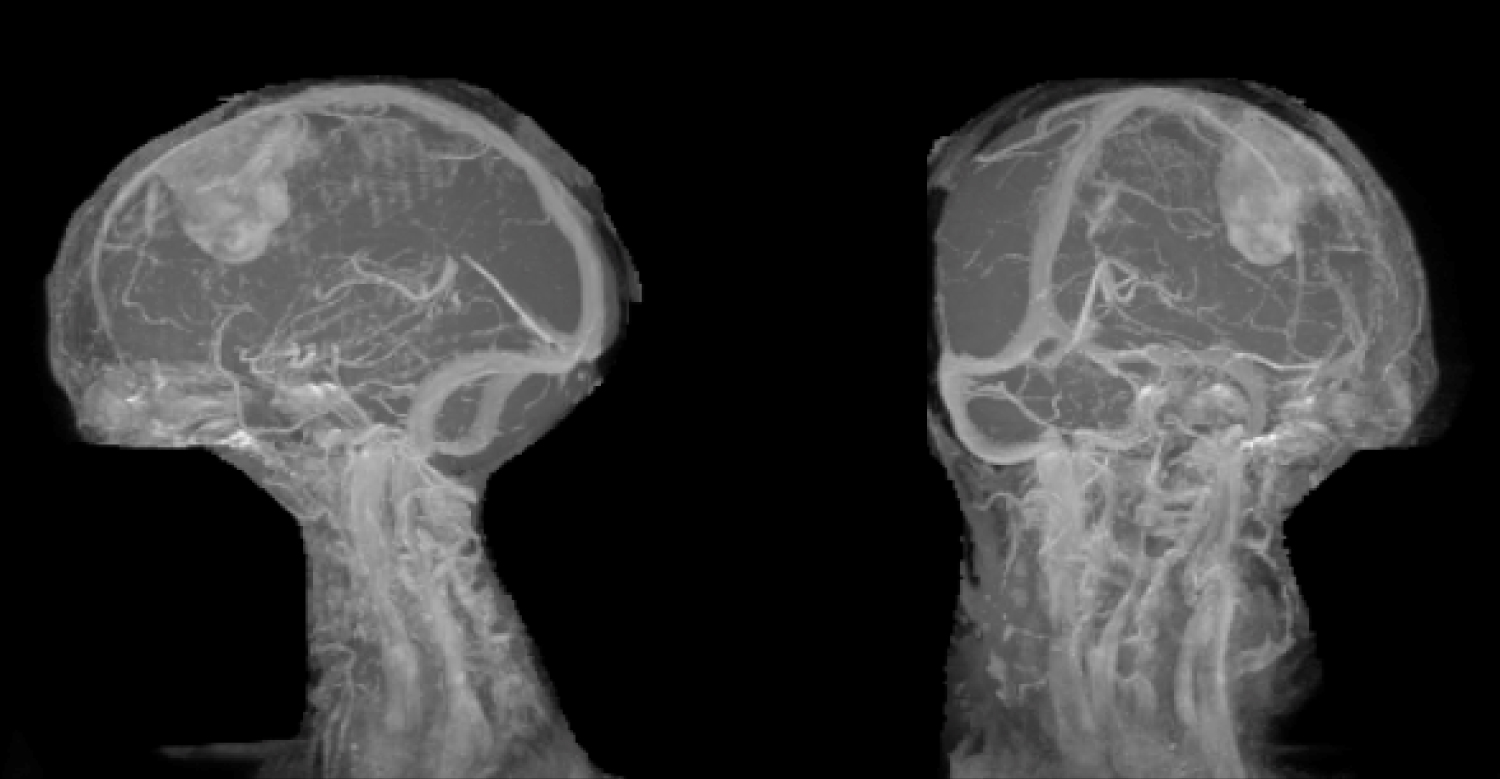 Figure 4: MR Venogram showing the lesion away from the major venous Sinuses in Sagittal and Coronal views.
View Figure 4
Figure 4: MR Venogram showing the lesion away from the major venous Sinuses in Sagittal and Coronal views.
View Figure 4
Once the HPE report was available, the relatives were counselled regarding adjuvant therapy. A second opinion was also taken to ensure this surprising diagnosis was confirmed. Due to the advanced age and wishes of the patient and her relatives, adjuvant chemo-RT was not given.
After 4 months she was again brought to hospital with dullness of sensorium and worsening of the weakness of the left upper and lower limb. A CT brain done showed a recurrence with perilesional oedema. The patient was immediately started on antiedema measures and steroids. She improved marginally. Surgery was suggested but refused by the relatives, as was RT and other forms of palliation. She was taken home for comfort care and succumbed to her disease gradually after 4 months. (Total post-operative survival was approximately 8 months).
Histopathology revealed a circumscribed cellular tumor composed of sheets of loosely cohesive large epithelioid and rhabdoid appearing polygonal tumor (Figure 5) cells with eccentric nuclei. Cells showed pleomorphic nuclei with prominent nucleoli, brisk atypical mitoses and abundant pink cytoplasm with hyaline inclusions (Figure 6). Periphery of tumor showed focal vague whorled pattern with ovoid cells, tumor necrosis and inflammatory cells. The diagnostic possibilities of a glioblastoma and rhabdoid meningioma were given with advised for immunohistochemistry for confirmation. Further immunohistochemistry revealed tumor cells being positive for positive for GFAP and focally positive for vimentin and negative for CK, EMA and HMB-45 and a final diagnosis of glioblastoma was given.
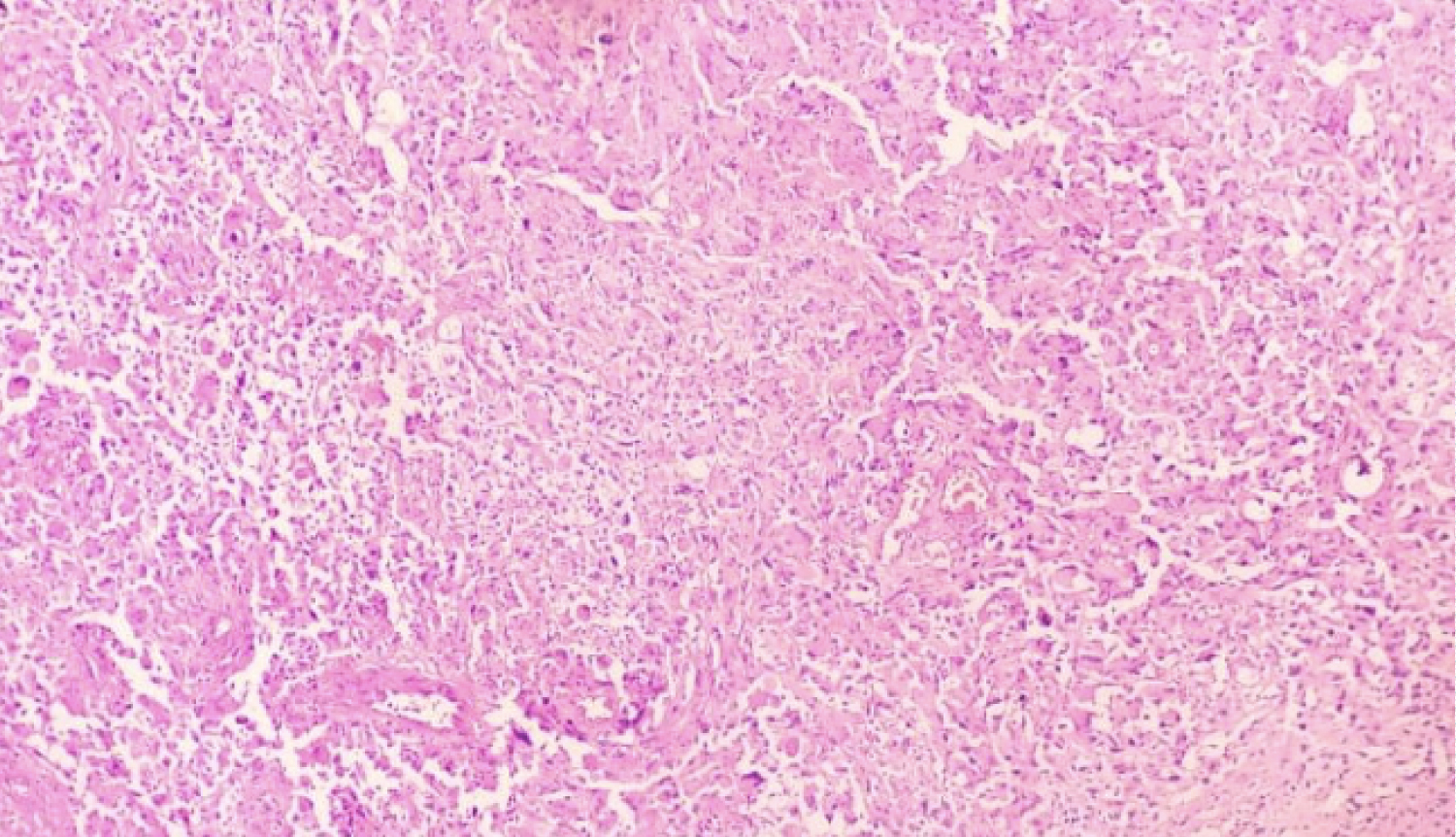 Figure 5: Highly cellular tumour with polygonal rhabdoid cells and foci of necrosis (H and E, × 20).
View Figure 5
Figure 5: Highly cellular tumour with polygonal rhabdoid cells and foci of necrosis (H and E, × 20).
View Figure 5
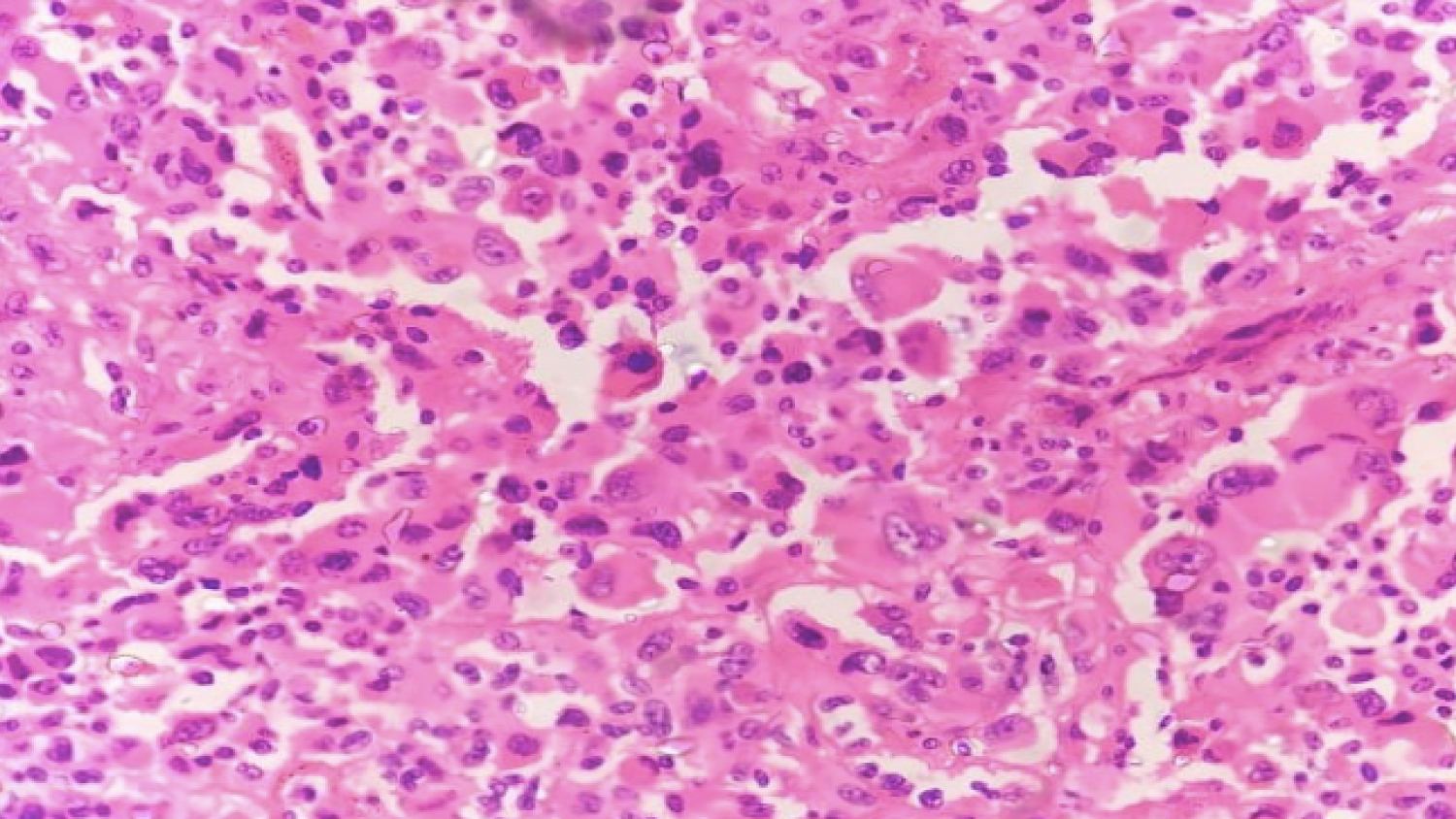 Figure 6: Large rhabdoid tumor cells with many heaving eccentric nuclei and abundant pink cytoplasm (H and E, × 40).
View Figure 6
Figure 6: Large rhabdoid tumor cells with many heaving eccentric nuclei and abundant pink cytoplasm (H and E, × 40).
View Figure 6
Glioblastoma with epithelioid and rhabdoid component on histology has been described in the literature but presenting as dura-based lesions are quite a rarity. In 2001, Wyatt-Ashmead found on histology that glioma had the structure of rhabdoid cells [1]. Based on this, they proposed the first case report and named this kind of disease as rhabdoid glioblastoma. Because the few numbers reported, in 2016, it was not got involved in the classification of Central Nervous System Tumor of WHO. Rhabdoid glioblastoma is a kind of highly malignant tumor that is common in young patients [1-3].
The predilection site locates at frontal and temporal lobe, with the characteristic of early recurrence and leptomeningeal spread. Differential diagnosis can be narrowed down with correlation with imaging findings.
Glioblastoma can mimic meningioma on magnetic resonance imaging with dural tail sign, CSF cleft sign and broad dural contact [4,5]. Histopathologically this was a high-grade tumor with predominantly epithelioid and rhabdoid morphology. First differential with this kind of cell morphology and imaging finding would be grade III meningioma with rhabdoid differentiation. In the central nervous system (CNS), most rhabdoid tumors occur in the posterior fossa of very young children and are associated with a primitive neuroectodermal tumor (PNET) component and are designated as atypical teratoid/rhabdoid tumors. Infrequently, other rhabdoid tumors of the CNS have been described, including rhabdoid meningiomas and malignant rhabdoid tumors of uncertain histogenesis [6,7].
On the aspect of histopathology, rhabdoid glioblastomas display tumor cells with round or oval shape, with cytoplasmic rich eosinophilic inclusions, eccentric nuclei with obvious nucleoli. In such cases final diagnosis can be arrived at by doing immunohistochemistry. Present case tumor cells showed positivity for GFAP and focal positivity for vimentin. IHC usually reveals that this kind of tumor shows GFAP and vimentin positive, focal expression of EMA, CK and INI-1 positive or special expression absence (Although Atypical teratoma/rhabdoid tumor, the cells show complete absence of INI-1 expression. While for Rhabdoid glioblastoma are GFAP positive with INI-1 expression is positive expression, giving concrete evidence to distinguish with above two diseases [7].
In 2016, Epithelioid glioblastoma [6], newly added in the CNS tumor classification of WHO shows characteristic cells with rich in eosinophilic cytoplasm and chromatin vacuoles and with large nucleoli. Parts of tumor cells have nuclear bias and para inclusion body, similar to Rhabdoid cells [1].
Histologically another close mimic is dural metastases by other malignant tumors. Most originate from cancers of the breast (34%), prostate (17%) and lung (13%), although others including bowel and germ cell tumors ependymomas, metastatic carcinomas, sarcomas and rhabdoid tumors.
Further confirmation can be done on immunohistochemistry. On imaging, they appear as focal nodular thickening of the dura, or when there is diffuse involvement, as smooth dural thickening following the contour of the calvarium [8]. Commonly used markers for confirmation are GFAP, CK, HMB-45 and showed diffuse positivity for D2-40 and vimentin. Focal positivity for EMA was also observed.
Even though there are very few cases of dura-based glioblastoma with rhabdoid features in literature, it should be kept in mind as a differential diagnosis with typical morphology. Final diagnosis can be arrived at, using relevant immunohistochemical markers.
No financial grants were received or used for this study.
No conflict of interest exists for the author.
Patient consent has been taken for this retrospective analysis.