

International Journal of Neurology and Neurotherapy
Introducing a Developed Model of Reversible Cardiac Arrest to Produce Global Brain Ischemia and Its Impact on Microtubule-Associated Protein Tau Phosphorylation at Ser396
Shohreh Majd1*, John H. Power2, Simon A. Koblar3 and Hugh J. M. Grantham1
1Neuronal injury and repair laboratory, Centre for Neuroscience and Paramedic Unit, School of Medicine, Flinders University, Adelaide, Australia
2Department of Human Physiology, School of Medicine, Flinders University, Adelaide, Australia
3School of Medicine, The Queen Elizabeth Hospital (TQEH) campus, University of Adelaide, Adelaide, Australia
*Corresponding author:
Shohreh Majd, Neuronal injury and repair laboratory, Flinders Medical Centre, Level 4, Rm 4D206, Bedford Park, SA 5042, Australia, E-mail: shohreh.majd@flinders.edu.au
Int J Neurol Neurother, IJNN-3-040, (Volume 3, Issue 1), Original Article; ISSN: 2378-3001
Received: January 29, 2016 | Accepted: February 24, 2016 | Published: February 26, 2016
Citation: Majd S, Power JH, Koblar SA, Grantham HJM (2016) Introducing a Developed Model of Reversible Cardiac Arrest to Produce Global Brain Ischemia and Its Impact on Microtubule-Associated Protein Tau Phosphorylation at Ser396. Int J Neurol Neurother 3:040. 10.23937/2378-3001/3/1/1040
Copyright: © 2016 Majd S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Objective: Brain ischemia is a consequence of stroke and cardiac arrest (CA), leading to short and long-term neurological impact involving cognitive function as well as dementia. An accurate, simple and reproducible model of CA ischemia and reperfusion is valuable in assessing the response to ischemia and therapeutic interventions. In the current study the effectiveness of a reversible model of CA has been assessed through examining the brain response in expressing tau and hyperphosphorylated tau (p-tau) protein, one of the main hallmarks of Alzheimer's disease.
Method: In the current study we used a two phase alternating current (AC)-derived reversible CA to generate global-brain ischemia through ventricular fibrillation (VF)/ventricular standstill followed by defibrillation, mechanical ventilation and standard resuscitation in 26 female adult Sprague Dawley rats. This model mimics CA in humans and allows testing of experimental interventions to restore/preserve brain function. Two phases of AC (24 Volts (V) followed by 18 V) were applied through a two-ring oesophageal wire. After 2-, 4- and 8-minute CA, resuscitation was initiated by ventilation, 8 Joules (J) chest electrical shock in the event of VF, adrenaline injection and manual chest compression.ECG, pulse rate and blood oxygen saturation were recorded during the resuscitation. Tau and p-tau levels were assessed as an indicator of neuronal response to ischemia.
Results: Resuscitation outcomes were assessed at 1st and 2nd hour and 4th week of restoration of blood circulation and animal survival. We report an early tau dephosphorylation which was followed by hyperphosphorylation after 4 weeks, while the total tau remained unchanged.
Conclusions: This method successfully produced reproducible global brain ischemia generating tau dephosphorylation in short-term recovery and hyperphosphorylation in long-term recovery. The findings show the efficiency of this technique in providing a tool to study the further neuronal response to ischemic situations and the underlying mechanisms.
Keywords
Global Brain Ischemia, Cardiac arrest, Reperfusion, Two phase alternating current, Tau phosphorylation.
Introduction
Stroke and cardiac arrest (CA) are not only the leading causes of death (approximately 800,000 case of stroke and 400,000 out of hospital CA per year in the U.S.) [1-3], but in the survivors of this ischemic injury, it is linked to an increased risk of cognitive and memory impairment [4,5] including the long-term effects of dementia [5]. In those who survive the initial CA nearly 60% die from neurological events and 30% of them suffer significant memory impairment [6]. In order to understand the pathophysiology of brain ischemia and suggest therapeutic approaches, there is a need for an accurate and reliable model of CA in which the variables such as ischemia time and resuscitation environment can be accurately controlled.
Interruptions to brain blood circulation have been achieved via various techniques including; asphyxia [7], compression of the heart vascular bundle by intrathoracic hook [8], potassium chloride (KCl) injection [9] and ventricular fibrillation (VF) [10,11]. Some procedures used to induce brain ischemia such as two-vessel occlusion (2-VO) with hypotension and four-vessel occlusion (4-VO) [12-14] are easy to perform, however they do not demonstrate all the features of the whole-body ischemia in a global ischemic situation such as CA. Other models of CA are complicated and invasive with little similarity to the actual situation of CA and reperfusion in a human. Inducing VF or ventricular stands till is more comparable to the human CA scenario and an appropriate model to mimic global brain ischemia after a complete CA [12]. Achieving CA, accurate control of ischemia time and control of the variable post resuscitation environment in an animal CA model is challenging due to the poor long-term animals' survival rates and complications associated with reperfusion.
Describing a reliable technique that avoids the previous limitations seems necessary. This technique could be used as a tool to improve the studies on mechanisms and effects of short-term and long-term global brain ischemia on brain. In the current study we hypothesized that two phases of intra oesophageal AC followed by cardiopulmonary resuscitation could generate a reversible model of global brain ischemia. Furthermore to verify the feasibility of this hypothesis and demonstrate the effectiveness of this method, investigating the early and long-term alteration of tau phosphorylation as one of the main brain responses to ischemia is the other purpose of this study.
Method
Animals and experimental groups
The animal experiments in this study were approved by the Animal Ethic Committee of Flinders University and were completed in accordance with the South Australian Prevention of Cruelty to Animals Act 1985 and followed the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 2004. Animals were divided to control group (anaesthesia only, no CA: n = 4) and ischemia groups (2 min CA, 2 min CA followed by 60 min reperfusion and 4 weeks recovery, 4 min CA followed by 60 min reperfusion, 8 min CA followed by 120 min reperfusion: n = 4 in each group).
Animal Preparation
Laboratory Animal Services of the University of Adelaide provided female Sprague-Dawley rats. The Animal Facility of Flinders University kept the rats (3 per cage) with free access to food and water until they reached two months of age and approximately 250-350 g. They were fasted for 12 hours before the experiments with free access to water. Anaesthesia was achieved using, intraperitoneal injection of Ketamine (Sigma, 343099) and Xylazine (Sigma, X1251), 100 mg/kg and 10 mg/kg body weight, respectively. Intravenous access was via 22G (0.90) mm intravenous catheter placed in a tail vein. Electrocardiogram leads attached to the right and left side of the chest allowed monitoring and defibrillation using a defibrillator/monitor (Philips HeartStart MRX, Philips Healthcare Inc., USA).The experimental time point is illustrated in Figure 1a. Oxygen saturation "(SPO2%) and pulse rate were monitored via a Pulse-oximeter. The optimal level of animal anaesthesia was confirmed through a toe and foot pinch method, as well as monitoring the respiratory pattern/rate and SPO2%.
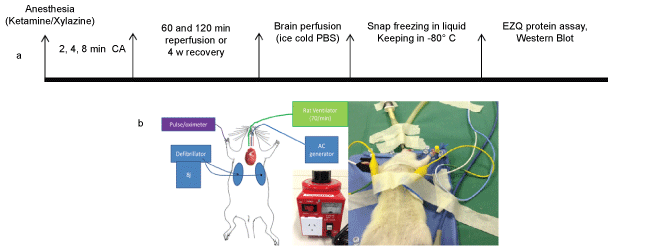
.
Figure 1: a) Experimental time lines of the study including the cardiac arrest (CA) durations, reperfusion and recovery periods, brain perfusion and separation, protein assessment and Western blot analysis. b) The position of the oesophageal pacing, connected to alternating current (AC) generator, defibrillating electrodes, connected to echocardiograph machine, intra-tracheal tube connected to the rat ventilator and pulse oximeter is described.
View Figure 1
Ventilation
Ventilation was performed via endotracheal intubation using a 16-gauge cannula inserted in the trachea and connected to a specific volume-controlled small animal ventilator (New England Medical Instruments Inc., Medway, Massachusetts, USA) with supplemental oxygen ventilating at 70 bpm with a tidal volume adjusted to 6 mL/kg.
Cardiopulmonary arrest protocol
To avoid extensive respiratory muscle paralysis associated with prolonged tetanic contraction and minimise thermal injury, we modified the previous method [15] by applying the two phase transoesophageal current. The alternating current (AC) 50 Hz AC 24 V (phase 1), followed by 50 Hz AC 18 V (phase 2) were delivered via a modified cardiac pacing wire (5F) with two end ring electrodes of 0.8 mm width and an inter-electrode gap of 4 mm, placed in the lower segment of the oesophagus (6-6.5 cm depth) posterior to the heart (Figure 1b), ensuring that the current was applied close to the heart without generating irreversible respiratory muscle paralysis. Mechanical ventilation was ceased for the duration of CA and a lack of output (pulse) was confirmed by the absence of pulse detected by the pulse oximeter at the same time that the AC current was recorded on the monitor.
Cardiopulmonary resuscitation protocol
We studied the effects of 2-, 4- and 8-minute CA followed by the post resuscitation/perfusion of 1hr, 2 hrs and 4 weeks. If indicated (VF/ventricular tachycardia post CA time) the rats were defibrillated (8J) via external electrodes connected to a defibrillator (Philips HeartStartMRx). Chest compressions at a rate of approximately 200-250/min and a depth of one third of the chest depth, with ventilations at a rate of 70 breaths per min and a volume of 6 mL per kilogram were delivered. Resuscitation was supported with intravenous adrenaline (0.01- 0.03 mg/Kg) and normal saline fluid boluses (1 ml) as needed. Effective resuscitation output and oxygen levels were confirmed by a pulse oximeter reading which relies on a pulse through a capillary bed to read SPO2%. Ventilation was started immediately via a small animal ventilator (New England Medical Instruments Inc., Medway, Massachusetts, USA). Restoration of perfusion was defined as the return of pulse detected by the pulse oximeter showing a mean SPO2% of 85-95% or greater during the time of resuscitation after CA. The animals were resuscitated and monitored for 1hr, 2 hrs and 4 weeks at recovery and the physiological scores were recorded based on the parameters described in Table 1. The animals were sacrificed painlessly under general anaesthesia after 2 min CA (2 min CA only group), 1 and 2 hours of resuscitation (2-, 4- and 8-minute CA followed by f 1 hr, 2 hrs resuscitation) and 4 weeks at recovery (2 min CA followed by 4 weeks at recovery). The brains were perfused with cold Phosphate Buffered Saline (PBS) and were collected. The specimens were frozen in liquid nitrogen immediately after isolation and kept in -80°C freezer for the further analysis.
![]()
Table 1: The physiological parameters of HR and SPQ2%, baseline and during the CA/CPR procedure, in control (anaesthesia only, no CA) and ischemia groups (I-2': 2 min ischemia, 2'/60': 2 min ischemia followed by 60 min reperfusion, 4'/60': 4 min ischemia followed by 60 min reperfusion, 8'/120': 8 min ischemia followed by 120 min reperfusion, 2'/4 w: 2 min ischemia followed by 4 weeks recovery period). HR: heart rate, SPQ2%: oxygen saturation percentage, CA: cardiac arrest, CPR: cardiopulmonary resuscitation.
View Table 1
Antibodies
Tau mouse monoclonal antibody (A-10; sc-390476) and phosphorylated tau rabbit polyclonal antibody (Ser396; sc-101815), were purchased from Santa Cruz. β-Actin mouse antibody (A2228) was purchased from Sigma. HRP donkey anti-mouse (715-036-150) and HRP donkey anti-rabbit (711-035-152) (secondary antibodies) were purchased from Jackson Immunoresearch (West Grove, USA).
Western blot analysis
The middle 1/3 (0.3-0.35 g) of the frozen brain specimens containing parietal cortex and hippocampus was homogenized in homogenizing extraction buffer containing protease inhibitors of Pepstatin A (Sigma, P5318, 1 μg/ml), Leupeptin (Sigma, L2884, 1 μg/ml) and phenylmethylsulfonyl fluoride (PMSF) (Sigma, P7626, 100 mM). The homogenate was centrifuged at 1000 × g for 5 min at 4°C and the supernatants were stored at -80°C until analysed.
The amount of total protein in each sample was calculated using an EZQ assay. To analyse electrophoretic mobility of p-tau, tau and β-actin, 30 μg of each sample in sample buffer was loaded in each well of AnykD™ TGX Stain-free gel (Bio-Rad, CA, USA, 569033). Samples were blotted on nitrocellulose membranes using a BioRad Trans-Blot transfer system kit (BioRad, CA, USA) according to the manufacturer's instructions. After electroblotting, the membranes were blocked for 1 hr at room temperature in a solution of 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 (pH 7.6). The separate membranes were incubated overnight at 4°C with primary antibodies of p-tau polyclonal antibody at Ser396 (1:250), tau monoclonal antibody (1:250), and β-actin (1:1800). On the following day the membranes were incubated for 1 hr at room temperature with the HRP secondary antibodies (donkey anti mouse, 1:3000; donkey anti rabbit, 1:3000). The blots were then developed using an ECL and chemiluminescence signal detection was performed using Fuji LAS4000 imager and quantitated by Care Stream molecular imaging software.
Statistical analysis
All of the data in the current study were analyzed using IBM SPSS Statistics version of SPSS Software and are expressed as the mean ± SEM. One-way ANOVA test was used to assess the differences between the means of the groups followed by post hoc Tukey's. Significance was defined as p < 0.05.
Results
Different durations of 2-, 4- and 8-minutecardiac arrest (CA) were tested followed by reperfusion. Reperfusion was achieved by effective resuscitation followed by return of spontaneous circulation where resuscitation was successful. Reperfusion times studied were 1 hr, 2 hrs and 4 weeks. The total mortality rate was 2 rats out of 26 due to respiratory arrest before applying the AC current. There were no differences between the control (anaesthesia only with no CA) and CA rats in respect to the baseline pre-arrest mean heart rate (HR) and SPO2% (Table 1). Before the induction of CA, all animals presented a normal sinus rhythm with the HR of 320 ± 30/min (Figure 2a). Applying AC stimulation caused a loss of pulse leading to an absence of pulse oximeter reading and a very obvious colour change in the capillary beds and eyes. During resuscitation physiological parameters including heart/pulse rate and SPO2% differed from the control group which underwent anaesthesia only with no CA (Table 1). Following the prescribed period of CA a number of different initial ECG rhythms were observed (Table 2) as follows:
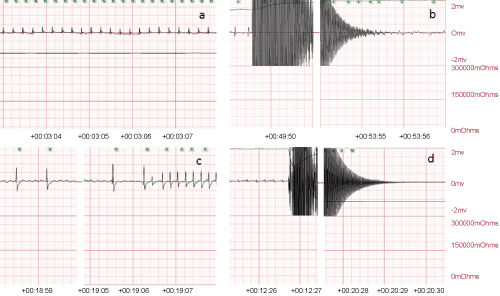
.
Figure 2: a) Normal sinus rhythm before applying the alternating current (AC). b) Ventricular fibrillation after 4 min delivering the high frequency current (24 V, 2 min followed by 18 V, the second 2 min). c) Sinus bradycardia following defibrillation and CPR which was improved to normal sinus rhythm. d) Asystolic rhythm after 8 min of CA.
View Figure 2
![]()
Table 2: ECG outcome by the time of AC termination and during cardiopulmonary resuscitation (CPR), in CA-treated (2, 4 and 8 min CA followed by 1 and 2 hrs CPR, 2 min CA followed by 4 weeks recovery) rats.
View Table 2
Ventricular fibrillation (VF) and asystole
The initial rhythm following AC current administration was ventricular fibrillation (VF) in 9 rats (Figure 2b) with no spontaneous reversion to sinus rhythm in 3 of them when cardiac resuscitation was terminated. VF in 6 rats responded to defibrillation (on one or more occasion) and CPR, generating sinus bradycardia in 4 rats, and normal sinus rhythm in 2 rats. Thesinus bradycardia improved to normal sinus rhythm after CPR (Figure 2c). Eight-minute CA was associated with an asystolic rhythm in 2 rats when the AC current was terminated (Figure 2d). Resuscitation involved defibrillation, CPR and IV adrenaline injections, in a process modelled on human resuscitation and attained an average SPO2 of 85 ± 10% with an average generated pulse of 200 ± 40 per min for up to 1 or 2 hrs (Table 1).
Normal sinus rhythm and sinus bradycardia
After applying the AC current 4 rats demonstrated an initial post current rhythm of normal sinus rhythm (Figure 3a). In this group, four rats had 2 min of CA. During resuscitation, the animal's average SPO2% reached 85 ± 10% and the generated pulse was 200 ± 40 per min. Upon termination of the AC current, 5 rats were in a bradycardia (Figure 3b), with one obvious AV block. These rhythms improved to normal sinus rhythm in 2 rats with resuscitation.
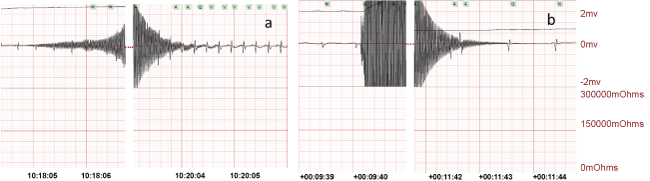
.
Figure 3: a) Normal cardiac sinus rhythm and b) sinus bradycardia after 2 min CA of 2 phases of high frequency transoesophageal AC (1 min 24 V followed by 1 min 18 V).
View Figure 3
Effect of CA and CA/reperfusion on tau phosphorylation (Ser396)
The effectiveness of our CA/reperfusion model was tested by assessing tau response to ischemia. Parietal cortical and subcortical hippocampus homogenates were used for Western blot analyses. In the ischemia only group, p-tau (Ser396) decreased (*p < 0.05, ischemic vs control, One Way ANOVA, followed by Tukey HSD Test). Similarly, p-tau (Ser396) also decreased in the 2-and 4-minute CA followed by 60 min reperfusion (*p < 0.01, ischemic/ reperfusion groups vs control, One Way ANOVA, followed by Tukey HSD Test). Tau showed hyperphosphorylation after 4 weeks recovery which was significantly different from the control, ischemia only and ischemia followed by 1 and 2 hr reperfusion groups (*p < 0.05, ischemic and 4 weeks recovery group vs control, One Way ANOVA, followed by Tukey HSD Test). Equal loading of each well was confirmed with similar β-actin levels across all lanes (Figure 4).
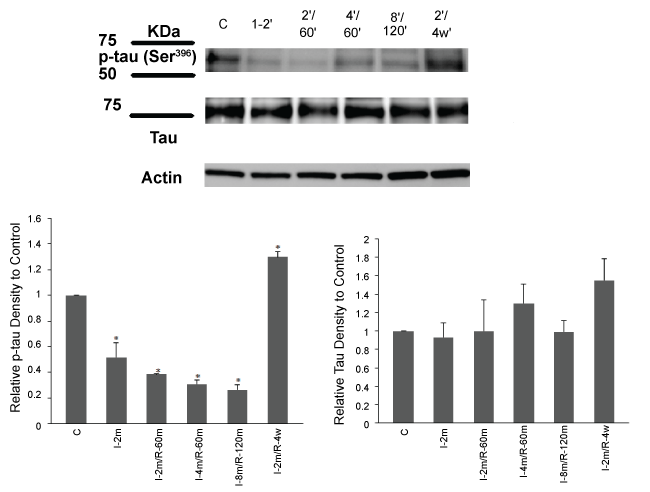
.
Figure 4: Tau phosphorylation reduced after 2 min ischemia, 2 and 4 min ischemia followed by 60 min reperfusion and 8 min ischemia followed by 2 hrs reperfusion. Tau showed a significant hyperphosphorylation after 4 weeks reperfusion. Total tau showed the similar levels in all groups. I-2': 2 min ischemia, 2'/60': 2 min ischemia followed by 60 min reperfusion, 4'/60': 4 min ischemia followed by 60 min reperfusion,8'/120': 8 min ischemia followed by 120 min reperfusion, 2'/4 w: 2 min ischemia followed by 4 weeks recovery period. One-way ANOVA, Data are expressed as mean ± SEM.*p < 0.01 ischemic/reperfusion groups vs control.*p < 0.05 ischemic and 2 min ischemia followed by 4 weeks recovery groups vs control.
View Figure 4
Effect of CA and CA/reperfusion on total tau expression
To examine any underlying changes in tau protein, we evaluated the level of total tau in the brain samples of the parietal cortical and subcortical hippocampus homogenates of the ischemia only group and the groups with different periods of ischemia and reperfusion (One Way ANOVA, ischemic and reperfusion groups vs control). The results showed that ischemia (2 min) and ischemia/reperfusion (2 and 4 min/1 hr; 8 min/2 hr; 2 min/4 weeks) did not affect the total tau protein expression while its phosphorylation was affected (Figure 4).
Discussion
In this study we developed a cardiac standstill model of CA by applying two declining voltages of AC current to produce a CA without irreversible respiratory muscle paralysis. Application of an AC current produced CA by causing a tetanic contraction of the heart muscle. Because of the effects of the AC current on cardiac function even if sinus rhythm persisted, manual compressions were always started at the end of the CA period giving an accurate reproducible start of reperfusion time. This is a reliable animal model with physiological similarity to the global ischemic situation in human CA allowing us to examine the effect of therapeutic approaches to CA and stroke which are the main causes of ischemic brain damage affecting millions of individuals around the world [16,17].
Many of the previous studies on brain ischemia/hypoxia have been performed using vessel occlusion techniques, such as occlusion of carotid arteries with or without vertebral and/or subclavian artery occlusion [12,13]. Apart from the invasive and irreversible nature of the models, the pathological and clinical effects of the global brain ischemia especially due to CA may differ from these situations and could have different influences on neurological outcome associated with this condition. VF/standstill is a technique that produces an absolute cessation of cerebral blood flow and perfusion of the other organs. This technique has been used in our study to mimic the clinical pathological condition of CA followed by the cardiopulmonary resuscitation (CPR). This animal model of CA represents a practical means of conducting experimental studies of CA and CPR. The importance of reducing the voltage in two phases of AC is related to the prolonged paralysis of respiratory muscles seen in animals with high non-reducing voltage current. Our model is one of the very few ones that used this technique in small animals to achieve an accurately controlled global brain ischemia and optimum reperfusion environment. We used defibrillation on animals that did not show an automatic return to normal cardiac rhythm at the time of AC termination and were in VF. As in human resuscitation, defibrillation after VF was accompanied with a combination of chest compressions (180-250 pm) and administration of adrenalin (0.01 mg/ kg increased to 0.03 mg/Kg if not responsive). Humans in CA demonstrate a similar range of rhythms to those demonstrated following AC current in our model [18]. In the present study, applying a high-frequency AC current of a decreasing voltage for a longer duration reduced the incidence of spontaneous reversion, and increased the incidence of VF. In no instance was there spontaneous reversion to normal sinus rhythm when the AC current persisted for 240 seconds or longer (480 seconds). The animals had a generated pulse in normal range of 160-240 bpm and SPO2% of 85-95%, during the time they were under resuscitation (short term for up to 2 hours). This arrest/resuscitation model offers advantages in terms of precision, minimal invasive features and simplicity to those wishing to study the effects of global ischemia in the short and long-term.
Low voltage current is not always able to generate VF, in small mammal hearts with very short refractory periods while high voltage current could be damaging [19,20]. This model achieves CA (cardiac standstill) without the necessity of achieving VF. It is been suggested that CA due to electrical accident is lethal in many cases because of the irreversible respiratory arrest leading to a hypoxic-derived secondary CA that occurs as a result of stiff respiratory muscles after contact with high voltage AC current [21,22]. Using a two phase gradually declining current delivered via the oesophagus decreases the risk of damage to respiratory muscles, associated with a constant high voltage current for the same period of time, making long-term recovery a viable option.
In the current study, we examined the microtubule associated tau protein and its phosphorylation level to test the efficiency of our model in generating brain ischemia. In recent years many models have been developed to study global ischemia; however some of them haven't reported histological changes in the brain tissue within the first 5-10 min [23,24]. Our results demonstrated an early tau dephosphorylation (Ser396), which was consistent with a stroke study, reporting tau dephosphorylation in a transient ischemic model of stroke [25]. In an animal model of 15 min ischemia, it has been shown previously that tau phosphorylation was reversible when the circulation was restored [26]. Our data showed no tau rephosphorylation at Ser396 occurred within the next hour of reperfusion following 2 min of CA, which suggests the involvement of a different mechanism responsible for altering p-tau status in shorter episodes of global brain ischemia. Our study showed that tau hyperphosphorylation at Ser396 increased significantly following 4 weeks recovery from CA, which was consistent with previous reports from animal models of ischemic brain injury showing tau protein phosphorylation following brain ischemia [26,27]. These significant changes in tau phosphorylation status demonstrating specific different responses to different times of ischemic insult and reperfusion, confirm that our model not only achieved ischemia, but also accurately controlled ischemic incident and reperfusion.
Conclusion
We suggest that development of VF/standstill model of CA in rats by our method of AC current delivered via the oesophagus is an applicable method without irreversible paralysis of respiration. This model is simpler when compared with the models of CA through AC current delivered directly to the endocardium of the right ventricle [20]. This method minimises the possible damaging effect of current producing epithelial thermal injury at the site of electrode placement, as well as to the heart, which was one of the disadvantages noted in some studies [28]. The pattern of p-tau alteration suggests that effective neuronal ischemia was generated by our model, with the tau hyperphosphorylated outcome, which is also seen in the human brain following ischemic stroke. This technique could be used in global ischemic studies with practical significance for facilitating experimental investigation in brain response to CA and resuscitation.
Conflict of Interest
There are no conflicts of interest to disclose in this study.
Acknowledgements
This study was supported by the Australian Resuscitation Council and a Flinders University Establishment grant. The authors would like to thank the Biomedical Engineering Department of Flinders University especially Mark McEwen and Noel Kitto for their technical support, the Animal Facility and Proteomics Facility of Flinders University, especially Dr. Timothy Chata way, and from the Alzheimer's and Parkinson diseases' lab especially Fariba Chegini for their enthusiastic discussions and support.
All authors involved in the study design and development of the model. SM and HG were responsible for completing the experiments. The initial draft was written by SM and reviewed by HG and JP.
References
-
Bunch TJ, White RD (2005) Trends in treated ventricular fibrillation in out-of-hospital cardiac arrest: ischemic compared to non-ischemic heart disease. Resuscitation 67: 51-54.
-
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, et al. (2011) Heart disease and stroke statistics--2011 update: a report from the American Heart Association. Circulation 123: e18-18e209.
-
Flynn RW, MacWalter RS, Doney AS (2008) The cost of cerebral ischaemia. Neuropharmacology 55: 250-256.
-
Pluta R, Ułamek M, Jabłoński M (2009) Alzheimer's mechanisms in ischemic brain degeneration. Anat Rec (Hoboken) 292: 1863-1881.
-
Pluta R (2002) Astroglial expression of the beta-amyloid in ischemia-reperfusion brain injury. Ann N Y Acad Sci 977: 102-108.
-
Herlitz J, Andersson E, Bång A, Engdahl J, Holmberg M, et al. (2000) Experiences from treatment of out-of-hospital cardiac arrest during 17 years in Göteborg. Eur Heart J 21: 1251-1258.
-
Jia X, Koenig MA, Venkatraman A, Thakor NV, Geocadin RG (2008) Post-cardiac arrest temperature manipulation alters early EEG bursting in rats. Resuscitation 78: 367-373.
-
Kawai K, Nitecka L, Ruetzler CA, Nagashima G, Joo F, et al. (1992) Global cerebral ischemia associated with cardiac arrest in the rat: I. Dynamics of early neuronal changes. Journal of cerebral blood flow and metabolism 12: 238-249.
-
Kofler J, Hattori K, Sawada M, DeVries AC, Martin LJ, et al. (2004) Histopathological and behavioral characterization of a novel model of cardiac arrest and cardiopulmonary resuscitation in mice. J Neurosci Methods 136: 33-44.
-
Padosch SA, Popp E, Vogel P, Böttiger BW (2003) Altered protein expression levels of Fas/CD95 and Fas ligand in differentially vulnerable brain areas in rats after global cerebral ischemia. Neurosci Lett 338: 247-251.
-
Popp E, Padosch SA, Vogel P, Schabitz WR, Schwab S, et al. (2004) Effects of intracerebroventricular application of brain-derived neurotrophic factor on cerebral recovery after cardiac arrest in rats. Crit Care Med 32: S359-S365.
-
Ginsberg MD, Busto R (1989) Rodent models of cerebral ischemia. Stroke 20: 1627-1642.
-
Yamaguchi M, Calvert JW, Kusaka G, Zhang JH (2005) One-stage anterior approach for four-vessel occlusion in rat. Stroke 36: 2212-2214.
-
Kovalska M, Kovalska L, Tothova B, Mahmood S, Adamkov M, et al. (2015) Combination of hyperhomocysteinemia and ischemic tolerance in experimental model of global ischemia in rats. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society 66: 887-897.
-
Chen MH, Liu TW, Xie L, Song FQ, He T, et al. (2007) A simpler cardiac arrest model in rats. Am J Emerg Med 25: 623-630.
-
Jabłoński M, Maciejewski R, Januszewski S, Ułamek M, Pluta R (2011) One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol Res 60 Suppl 1: S113-119.
-
Mateen FJ, Josephs KA, Trenerry MR, Felmlee-Devine MD, Weaver AL, et al. (2011) Long-term cognitive outcomes following out-of-hospital cardiac arrest: a population-based study. Neurology 77: 1438-1445.
-
Pokorný J, Staněk V, Vrána M (2011) Sudden cardiac death thirty years ago and at present. The role of autonomic disturbances in acute myocardial infarction revisited. Physiol Res 60: 715-728.
-
Song L, Weil MH, Tang W, Sun S, Pellis T (2002) Cardiopulmonary resuscitation in the mouse. J Appl Physiol (1985) 93: 1222-1226.
-
Dave KR, Della-Morte D, Saul I, Prado R, Perez-Pinzon MA (2013) Ventricular fibrillation-induced cardiac arrest in the rat as a model of global cerebral ischemia. Transl Stroke Res 4: 571-578.
-
Seidl S (2006) Pathological features of death from lightning strike. Forensic Pathology Reviews 4: 3-23.
-
Spies C, Trohman RG (2006) Narrative review: Electrocution and life-threatening electrical injuries. Ann Intern Med 145: 531-537.
-
Kawahara N, Kawai K, Toyoda T, Nakatomi H, Furuya K, et al. (2002) Cardiac arrest cerebral ischemia model in mice failed to cause delayed neuronal death in the hippocampus. Neurosci Lett 322: 91-94.
-
Mizushima H, Zhou CJ, Dohi K, Horai R, Asano M, et al. (2002) Reduced postischemic apoptosis in the hippocampus of mice deficient in interleukin-1. J Comp Neurol 448: 203-216.
-
Mailliot C, Podevin-Dimster V, Rosenthal RE, Sergeant N, Delacourte A, et al. (2000) Rapid tau protein dephosphorylation and differential rephosphorylation during cardiac arrest-induced cerebral ischemia and reperfusion. Journal of cerebral blood flow and metabolism 20: 543-549.
-
Song B, Ao Q, Wang Z, Liu W, Niu Y, et al. (2013) Phosphorylation of tau protein over time in rats subjected to transient brain ischemia. Neural Regen Res 8: 3173-3182.
-
Dong DW, Zhang YS, Yang WY, Wang-Qin RQ, Xu AD, et al. (2014) Hyperphosphorylation of tau protein in the ipsilateral thalamus after focal cortical infarction in rats. Brain Res 1543: 280-289.
-
Abella BS, Zhao D, Alvarado J, Hamann K, Vanden Hoek TL, et al. (2004) Intra-arrest cooling improves outcomes in a murine cardiac arrest model. Circulation 109: 2786-2791.
