Susac Syndrome is an infrequent condition that is often misdiagnosed as Multiple Sclerosis. This syndrome is characterized by the clinical trial of encephalopathy, retinopathy with branch retinal artery occlusions and hearing loss.
We describe a patient with Susac Syndrome that was initially diagnosed as having multiple sclerosis with clinical deterioration after starting treatment with interferon beta-1a.
Despite the classic triad of encephalopathy, branch retinal occlusions and hearing loss is pathognomonic of Susac Syndrome, it is present in only a small percentage of patients. In our case, at the onset of symptoms the triad was incomplete and thus the correct diagnosis and treatment were delayed.
Although the clinical trial was incomplete, our patient had the classic magnetic resonance appearance with branch retinal artery occlusions and sensorineural hearing loss. The cerebrospinal fluid examination was normal with no oligoclonal bands and a no elevated IgG index.
Our patient responded well to the correct immunosuppressive treatment, with clinical improvement. The lesions in the magnetic resonance also improved after the treatment.
Susac syndrome must be considered in the differential diagnosis of MS, especially in cases with suggestive symptoms, lesions involving corpus callosum, no oligoclonal bands in LCR and in patients with progressive worsening despite a correct DMT. A prompt diagnosis is essential in order to prevent disability or irreversible sequelae related to the disease.
Susac syndrome, Demyelinating diseases, Multiple sclerosis
ScS: Susac Syndrome; MS: Multiple Sclerosis; MRI: Magnetic Resonance; CSF: Cerebrospinal Fluid; MTP: Methylprednisolone; IFN b1a: Interferon beta-1a; BRAO: Branch Retinal Arteriolar Occlusions; MM: Mycophenolate Mofetil
Susac Syndrome (ScS) is an infrequent condition with unclear pathophysiology. The most suspected pathogenetic mechanism is an autoimmune microangiopathy, affecting the precapillary arterioles of the brain, inner ear and retina [1,2]. The syndrome has been defined by a specific triad of symptoms: Encephalopathy with focal neurological signs (that may be associated to a new headache, no more than 6 months before the onset of the latter symptoms); branch retinal arterial occlusions and hearing loss. The classic triad is considered pathognomonic, however, it is only present in 13% of patients, leading to a delayed or wrong diagnosis in many cases [3]. Multiple Sclerosis (MS) is the most common misdiagnosis of ScS, and the immune treatment for MS can exacerbate the disease [4,5].
We report the case of a patient with ScS that was initially diagnosed with MS. We describe the investigative findings that differentiate ScS from MS and the clinical course after first-choice treatment.
A 46-year-old woman with no relevant medical history, was referred to our multiple sclerosis department with a two years history of MS with no response to immunosuppressive treatment.
The first symptoms started two years before with dizziness and a facial palsy at the onset. She underwent a cerebral Magnetic Resonance (MRI) showing T2-weighted lesions, that were suggestive of multiple sclerosis (Figure 1). A lumbar puncture showed a normal Cerebrospinal Fluid examination (CSF) with no oligoclonal bands and no elevated IgG index. The initial diagnosis was remittent-recurrent MS, and she was treated with intravenous Methylprednisolone (MTP) pulses during five consecutives days, following with Interferon (IFN b1a) as the usual Disease Modifying Treatment (DMT). The symptoms did not improve. After two months she experienced sudden loss of vision in the left eye, so she was evaluated by an ophthalmologist. A visual field examination revealed a superior altitudinal defect in the left eye and a fundoscopy showed findings suggestive of retinal vasculitis. She was treated with oral corticosteroids and after that she remained neurologically stable.
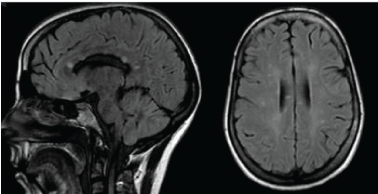 Figure 1: Sagittal and axial FLAIR images showing callosal and deep white matter hyperintense lesions. View Figure 1
Figure 1: Sagittal and axial FLAIR images showing callosal and deep white matter hyperintense lesions. View Figure 1
One year later, she was admitted through the emergency department due to unusual headache and aphasia, with weakness of the right arm and somnolence. A new cerebral MRI was done showing confluent T2-weighted lesions in the corpus callosum, basal ganglia and profound bilateral cerebral white matter (Figure 2) An electroencephalogram showed generalized diffuse slowing suggestive of encephalopathy. Treatment with 5 pulses of MTP was initiated with mild improvement and then she was treated with intravenous immunoglobulins (0.4 g/kg daily for five days) with significant improvement since its early days.
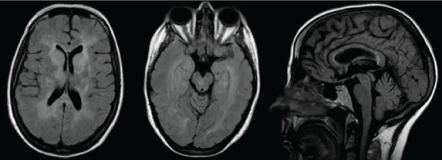 Figure 2: Axial and sagittal FLAIR images showing confluent lesions in the profound bilateral cerebral white matter and callosal hyperintense lesions. View Figure 2
Figure 2: Axial and sagittal FLAIR images showing confluent lesions in the profound bilateral cerebral white matter and callosal hyperintense lesions. View Figure 2
After that, she was referred to our multiple sclerosis department due to poor clinical evolution despite treatment.
During the first visit at our department, the patient complained of visual loss, tinnitus and dizziness. Findings on physical examination were decreased visual acuity, hearing loss, mild troncular ataxia and exaggerated deep tendon reflexes. A new cerebral MRI showed multiple small T2-weighted lesions involving corpus callosum and periventricular white matter lesions, with enhancement after gadolinium injection in some of them. Retinal fluorescein angiography showed multiple retinal arteriolar branch occlusions (BRAO) and an audiometric evaluation revealed bilateral low-frequency sensorineural hearing loss.
She was diagnosed of Susac Syndrome (ScS) and Mycophenolate Mofetil (MM) was started. On the follow up, the patient condition remained stable, but three months after the initiation of MM, the cerebral lesions improved (Figure 3).
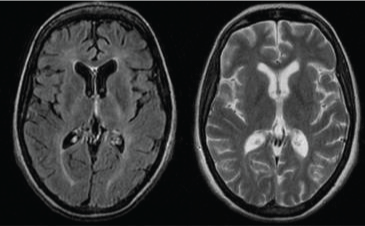 Figure 3: Axial T2-weighted images showing improvement of the cerebral MRI features compared to the previous cerebral MRI. View Figure 3
Figure 3: Axial T2-weighted images showing improvement of the cerebral MRI features compared to the previous cerebral MRI. View Figure 3
Susac Syndrome is a rare disease that affects predominantly young woman aged between 20-40 years [2,6]. The diagnosis is based on clinical and paraclinical findings in the brain, inner ear and retina.
The pathogenesis of ScS remains unclear, but it is considered due to an autoimmune microangiopathy with an inflammation-related occlusion of the microvessels in the brain, retina and inner ear. Other entities such viral infections, idiopathic vasospasm or hypercoagulable phenomena have been suggested as causes of ScS, but evidence is scant [6]. The findings of endothelial C4d deposition, lymphocytic perivascular infiltrates and circulating anti-endothelial cell antibodies in patients affected by this disease supports an autoimmune origin [2].
The MRI studies in patients affected by ScS revealed characteristics findings within the corpus callosum, with T2 weighted lesions that used to be well-demarcated, spherical and located preferentially in the central of the corpus callosum, with a "snowball" appearance [2]. The white matter is also frequently involved. These lesions are commonly mistaken with other demyelinating processes, typically MS.
CSF examination may show a mild elevation of total proteins and mild pleocytosis (usually less than 12 lymphocytes/mL) Oligoclonal bands or intrathecal IgG synthesis are rarely found [2,3].
The first choice treatment is currently unknown. Empiric treatment is based on the hypothesis of an autoimmune origin [3]. In the acute episode, treatment includes high-dose corticosteroids, followed by a continuous immunosuppressive therapy [3,7]. The most commonly used immunosuppressants are azathioprine, cyclophosphamide, mycophenolate mofetil, plasma exchange or intravenous immunoglobulins [7,8]. The clinical course is usually self-limited, lasting from months to years, and with varying residual disabilities. It remains unclear as to how much immunosuppressive treatment is required and for how long [7], however, in patients in whom treatment is delayed, permanent sequelae is higher [8,9]. Some authors also support the potential benefits of antithrombotic or anticoagulant therapy [2].
Regarding our patient, a relationship between the treatment with MM, the improvement on neuroimaging and the clinical stabilization was observed. Despite an initial unfavorable course, a clinical improvement was reported a few weeks after treatment started, and still remain. The effectiveness of treatment with MM supports an autoimmune origin of the disease.
ScS is a rare disease that must be considered in the differential diagnosis of MS, especially in cases with suggestive symptoms, lesions involving corpus callosum, no oligoclonal bands in LCR and in those patients with progressive worsening despite a correct DMT. Considering ScS is important from both a diagnostic and timely therapeutic standpoint. A prompt diagnosis is essential in order to prevent disability or irreversible sequelae related to the disease.