

International Journal of Pediatric Research
A New Outlook on Clinical Course of Juvenile Dermatomyositis - Experience of a Single Center
Krzysztof Orczyk, Joanna Swidrowska, Jerzy Stanczyk and Elzbieta Smolewska*
Department of Pediatric Cardiology and Rheumatology, Medical University of Lodz, Poland
*Corresponding author: Elzbieta Smolewska, Department of Pediatric Cardiology and Rheumatology, Medical University of Lodz, Poland, E-mail: e.smolewska@wp.pl
Int J Pediatr Res, IJPR-1-004, (Volume 1, Issue 1), Research Article; ISSN: 2469-5769
Received: March 11, 2015 | Accepted: April 06, 2015 | Published: April 08, 2015
Citation: Orczyk K, Swidrowska J, Stanczyk J, Smolewska E (2015) A New Outlook on Clinical Course of Juvenile Dermatomyositis - Experience of a Single Center. Int J Pediatr Res 1:004. 10.23937/2469-5769/1510004
Copyright: © 2015 Orczyk K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction: Juvenile dermatomyositis (JDM) is the most common inflammatory myopathy in children. Diagnostic criteria by Bohan & Peter are originally formulated for adults. A wide range of differences in clinical course of dermatomyositis between adults and children has provoked to set a new look at the existing criteria.
Objective: The aim of our study was to analyze the clinical course and reaction to administered drug therapy in association with laboratory and electromyographic findings in pediatric patients with dermatomyositis.
Materials and methods: The retrospective analysis included 5 children with JDM hospitalized in Department of Pediatric Cardiology and Rheumatology Medical University of Lodz, Poland between October 2000 and November 2014. Demographic data, characteristics of clinical symptoms, laboratory tests, EMG and muscle biopsy reports and reaction to administered treatment were analyzed.
Results: Muscle weakness was the most common symptom observed in 80% of patients. Other symptoms present in more than a half of patients included heliotrope rash (60%) and Gottron sign (60%). Muscle enzyme levels were significantly elevated in 4 out of 5 patients. Electromyography (EMG) was performed in 3 patients and was positive in every case, as well as muscle biopsy. Initial therapy involved prednisone 1mg/kg b.w./day in all patients combined with disease modifying anti-rheumatic drug, whether methotrexate was the most common choice (60%). Complications which occurred in presented patients involve: rhabdomyolysis, severe calcinosis and contractures of elbows, exacerbation after reduction of GCS dose, hypertriglyceridemia, steatohepatitis, tachycardia.
Conclusion: Our study reports remarkable characteristics of clinical course of JDM. Further analysis needs to be performed in order to adjust diagnostic criteria to pediatric patients.
Introduction
Juvenile dermatomyositis (JDM) is a rare systemic disease that involves principally the musculoskeletal and cutaneous systems [1]. As JDM affects 2-3 children per million per year [2], it remains the most common inflammatory myopathy in pediatric patients [3].
Classical JDM manifests with progressive muscle weakness, easy fatigue, skin rash and fever [4]. Nonetheless, its course varies from mild muscle symptoms that are easy to treat and never relapse, to a therapy-resistant chronic condition [5].
JDM diagnosis is based on the Bohan & Peter criteria [6] originally developed for adult patients: symmetric proximal muscle weakness, biopsy-proven myositis, elevated serum muscle enzyme levels, electromyographic changes of myositis. JDM may be also misdiagnosed as polymyositis in patients presenting with isolated muscle symptoms on first admission [7].
The pathogenesis of JDM is not completely understandable yet.Viral infection or immune dysfunction may trigger disease in patients with genetic predispositions [1]. Accordingly, the identification of novel autoantibodies in JDM (such as anti-p155/140, anti p140) may have clinical implications, as they are associated with specific clinical features, treatment response and prognosis [8].
Dermatomyositis is also considered as one of paraneoplastic syndromes in adults. It is believed to occur in 7-15% of all cancer patients [9]. The association of dermatomyositis and cancer is more likely to happen in elderly people [10], but it may also coincide with leukemia and lymphoma in pediatric patients [11].
As JDM differs from the course of disease in adults, diagnostic criteria require a new approach. They do not consider radiologic methods such as MRI, which is recently becoming the preferred non invasive test indicating muscle inflammation, instead of muscle biopsy and electromyography (EMG) included in the criteria [3].
In order to adjust criteria to pediatric patients, differences in clinical course of dermatomyositis between adults and children need to be reported. Therefore we present series of 5 cases.
Materials and Methods
The retrospective study included the medical charts of 8 patients ≤18 years of age who were treated due to JDM suspicion at the Department of Pediatric Cardiology and Rheumatology, Medical University of Lodz, between October 2000 and November 2014. These charts contained patient's clinical presentations, results of laboratory tests and clinical outcomes. Total patient's history was studied in every case, not only initial hospitalizations. Three of patients who failed to be finally diagnosed with JDM were excluded from the study.
The following parameters were recorded: gender, age at diagnosis, duration of symptoms, initial manifestations, EMG and muscle biopsy reports, laboratory data, medication use, clinical complications. Duration of symptoms was defined as the time period from disease onset until diagnosis. Creatine kinase (CK), lactate dehydrogenase (LDH), erythrocyte sedimentation rate (ESR), the activity of aspartate and alanine aminotranspherase (AST, ALT) and presence of autoantibodies were particularly checked among the results of laboratory tests. Every patient was verified whether initial symptoms involved lesions characteristic for JDM such as patognomonic Gottron sign (erythematosus rash or scaly eruptions typically over the extensor joint surfaces), heliotrope rash accompanied by periorbital oedema [7].
The sample was too small to perform statistical analyses. Furthermore, the descriptive character was the original assumption of the study.
Results
Eight patients with suspected JDM were evaluated in the study. One patient was excluded due to final diagnosis other than JDM (Ascaris lumbricoides infection). Two were lost before the end of diagnostic process as they did not fulfill criteria during first hospitalization. One of them presented with muscle weakness, erythema of eyelids and macular rash on both upper and lower extremities, but after negative result of skin and muscle biopsy and non diagnostic EMG further diagnostic process was postponed. The latter was referred to the department with history of oedema of peripheral joints and itchy rash on whole surface of the skin after respiratory infection. Clinical manifestations and results of laboratory tests were within normal limits though, so the patient was recommended to present again in case of relapse of original symptoms.
In addition, one patient was initially diagnosed with polymyositis. After evaluation of total patient's history the patient was re-diagnosed with JDM and included in the study.
The clinical characteristics of 5 patients who were finally diagnosed with JDM are depicted in Table 1. Our series included 3 girls and 2 boys; the mean age on diagnosis was 4.6 years. Two girls presented after 4 months of symptomatic disease, whereas one boy was admitted 9 months after symptoms began. The two remaining patients were previously diagnosed in other centers and their first presentation to the department was caused by JDM exacerbation, therefore such data was unavailable to be obtained.
![]()
Table 1: Clinical characteristics of the patients finally diagnosed with JDM
View Table 1
Manifestations characteristic for JDM were frequent in the group: muscle weakness occurred in 4 out of 5 patients, heliotrope rash in 3, Gottron sign in 3, muscle pain in 2, fever in 2 (which in one case lead to suspicion of rheumatic fever). One patient complained of movement difficulties. There were no patients with Gottron papules though.
Besides, patients presented a number of symptoms different than mentioned above, like: lupus-like butterfly rash (which needed differential diagnosis with SLE); hair loss; macular rash on nose; scar-like lesions on hands (which probably were a consequence of developing calcinosis in this patient); fibromatous tumour in the posterior region of the neck (which needed to be consulted by a surgeon who recommended an oncologic control); pneumonia on first admission (which corresponds with environmental triggers theorem).
EMG and muscle biopsy was performed in 3 patients and gave positive results in every case it was performed. In one patient both EMG and muscle biopsy was omitted due to specific symptoms which were enough to make a diagnosis. Histopathological examination revealed muscle lymphocyte infiltration in 2 cases, whether in 1 case the perivascular lymphoid infiltration was additionally observed. Speaking of laboratory tests, detailed results are presented in Table 2. Summarizing, 4 out of 5 patients had elevated level of CK. The remaining patient was initially admitted with exacerbation as mentioned above; therefore it may be a false negative result.
![]()
Table 2: Detailed results of laboratory tests on first admission to the hospital
View Table 2
In 3 patients there were elevated levels of aspartate aminotranspherase AST and ESR. Two of them had also elevated level of LDH. In one case level of myoglobin was above the normal range as well (2125μg/L in patient #3). Increased level of total IgE was observed in 4 patients. Antinuclear antibodies (ANA) were present in all cases with PM-Scl antibodies found in patient #3.
Brief summary of medications used in each patient is included in Table 1.
Treatment in all patients was started from prednisone 1mg/kg b.w./day. Progressive reduction of dosage was managed in all cases from the first month of therapy. In 3 patients the decrease of doses was caused by positive response to the treatment, leading to discontinuation of prednisone in 2 cases (patient #1 and #3) after 4 years and 3 years and 8 months, respectively. In 2 other patients (#4 and #5) prednisone did not lead to clinical improvement and further medications had to be introduced, therefore prednisone dose was decreased in order to limit probable adverse GCS effects.
In 3 patients who presented in severe condition during first admission, prednisone administration was proceeded by intravenous methylprednisolone pulse therapy (20mg/kg b.w.). It was a one-time treatment for patient #3, whereas in 2 remaining cases the polycyclic course lasted for 2 years and 10 months (#4) and 3 years and 10 months (#5).
Glucocorticosteroid therapy was combined with disease-modifying anti-rheumatic drug (DMARD) in 4 patients. A first-line drug was preferably methotrexate (MTX) 10 mg/week (3 out of 4 cases). In patient #3 it remained the only treatment after discontinuation of prednisone. Two other patients continued MTX therapy for 5 years and 3 months (#4) and 6 years and 2 months (#5) until it was replaced by azathiopryne (AZA).
Hydroxychloroquine (HCQ) was added to the therapy in 2 cases: during the first hospitalization in patient #2 and 6 months after diagnosis in patient #4.
Biological treatment was initiated in 2 patients who did not respond for medications theretofore. Patient #4 was treated with infliximab (IFX) 3mg/kg b.w./dose for 10 months and then 6mg/kg b.w./dose for 7 months without clinical improvement. By contrast, patient #5 responded positively to adalimumab (ADA) and has been effectively continuing this treatment for 5 years until the end of observation period.
Intravenous immunoglobulin (IVIG) infusions were also administered in 2 aforementioned patients. For patient #4 it was the first effective treatment and a one-year therapy substantially improved his condition. In patient #5 it was introduced in order to rebalance the disease after rhabdomyolysis which he developed 4 years after the first hospitalization.
Patient #4 received also pamidronate (PAM) therapy which started 2 years and 10 months after diagnosis and lasted for 2 years and 10 months. It was introduced because of severe systemic calcinosis complicated with osteoporosis, which he developed along with contractures in elbows (Figure 1). Signs of calcinosis and contractures subsided during the one year IVIG therapy.
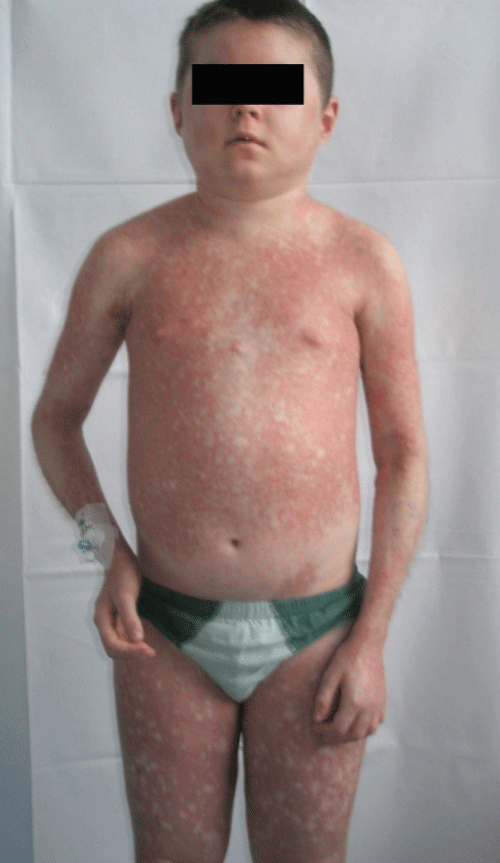
.
Figure 1: An 8-year-old JDM patient with severe calcinosis, erythematosus skin lesions and contractures in elbows.
View Figure 1
These and other complications which occurred in our patients are listed in Table 3.
![]()
Table 3: Complications
View Table 3
Discussion
Our evaluation of clinical course in 5 patients revealed both typical and uncharacteristic symptoms and complications of JDM. Gottron sign, which is described as patognomonic symptom of the disease, was noted in 3 cases. Characteristic manifestations, such as muscle weakness, heliotrope rash, muscle pain and fever, were also observed. Additional, non-specific symptoms found in analyzed patients included: lupus-like butterfly rash, hair loss, macular rash on nose, scar-like lesions on hands, fibromatous tumour in the posterior region of the neck, pneumonia on first admission. Findlay et al. [7] listed other symptoms occurring in patients with JDM: V sign (erythematosus macular rash on the face, neck and chest), shawl sign (when rash is located on the back of the neck and shoulders), hyperkeratosis, horizontal fissures on the palms, periungual teleangiectasias. These manifestations were not observed in our patients though.
Kumar et al. [3] resumed median duration of symptoms prior to diagnosis in a few studies, which varied from 2.8 to 9.25 months. It accounted for 4 months in our study.
Although Martin et al. [12] stated that JDM symptoms may depend on the age at diagnosis, we did not recognize such regularity. Cutaneous manifestations were present in patients both under and over 5 years old, whereas symptoms like headache or Raynaud's syndrome were not observed at all.
80% of patients in our report presented both muscular and cutaneous symptoms. Similarly, Sun et al. [13] revealed that 17,9% patients had only cutaneous manifestations without muscle weakness. On the contrary, Rider et al. [5] suggested the high frequency of photosensitivity in JDM patients, whereas it did not appear in our cases.
Calcinosis is not frequently present at diagnosis, but it is more commonly seen in juvenile patients and may be induced by delay in diagnosis [14]. Presley et al. [15] pointed out surgery, trauma or inflammation as trigger factors. According to our findings, severity of calcinosis may depend on the JDM activity. However, Meher et al. [16] reported a case of JDM who presented with extensive calcinosis long before development of rash and muscle weakness.
Sun et al. [13] reported that significant portion of patients has muscle enzyme levels within normal limits on diagnosis. It is not consistent with our study as 80% of patients had elevated levels of CK and the remaining patient might be false negative.
In accordance with Ishida et al. [17] who stated that JDM patients have significantly higher levels of total IgE, we observed such regularity in 4 patients.
As mentioned before, MRI is frequently performed during diagnostic process in place of EMG and muscle biopsy included in the criteria. Sanyal et al. [14] also suggested that radiography may be helpful in diagnosis as cutaneous signs in Indian population and early muscle weakness in an already sick child do not appear as reliable manifestations. Besides, Habers et al. [18] postulated usage of quantitative muscle ultrasonography in place of MRI, which necessity for sedation in children limits its common use.
Initial therapy of all patients included in the study involved prednisone 1mg/kg b.w./day. In 60% of patients it was preceded by intravenous methylprednisolone 20mg/kg b.w./dose. 60% of patients were treated with MTX 10mg/week. In one patient it was introduced during the first hospitalization whereas in two others it was added to the treatment after one month of GCS use. IVIG was introduced in 2 patients but not as a part of initial therapy. In one case it was preceded by ineffective IFX course. In the remaining case it was followed by effective ADA treatment.
In 2012 CARRA developed consensus protocols of JDM therapy [19]. All three treatment arms involve prednisone 2mg/kg b.w./day and MTX 1mg/kg b.w./week as the initial therapy. Intravenous methylprednisolone 30mg/kg b.w./day is also included in two treatment plans with additional IVIG 2g/kg b.w./dose in one of them. These protocols were aimed to limit adverse effects of high-dose GCS use.
Summary of our treatment approach differs from CARRA consensus as it was published in 2012 and there were no specific guidelines of treatment theretofore. According to the present knowledge patients included in our study may have been treated differently.
Identified adverse factors for disease remission included male sex and positive Gowers' sign [13]. Male patients reported in our study had more severe clinical course indeed. We did not observe Gowers' sign though.
Fujita et al. [10] reported two adult cases of interstitial pneumonia concomitant with dermatomyositis on diagnosis. One of patients in our study presented with pneumonia on first admission, and the other one needed a one-week break from MTX therapy due to respiratory tract infection. Marie [11] identifies that lung disease may be a direct consequence of muscle weakness and subsequent ventilatory insufficiency. Therefore relationship between dermatomyositis and pneumonia should be further investigated.
As mentioned before, JDM is usually not associated with cancer which concomitance is regular in adults. However, one of our patients needed to be referred to oncologist. Therefore neoplastic diseases are worth being excluded in JDM patients.
Patients included in our study were not tested for anti-p155/140 and anti-p140 antibodies due to its poor availability. Yu et al. [8] suggested also testing for antiendothelial cell antibodies (AECA), but its specificity is questionable.
Troyanov et al. [20] postulated the new criteria to differentiate pure dermatomyositis from overlap myositis with DM features. This manner of diagnostic process would not be applicable in our patients as characteristic rashes were not the first manifestation followed by muscle weakness in every case. Furthermore, patients diagnosed with JDM in our study had elevated level of CK despite the absence of DM-specific antibodies like anti Mi 2.
The small size of sample is the main limitation of our study. Although a group of 5 patients is not enough to draw profound conclusions, this study might provide appropriate data to be included in future meta-analysis in order to rethink the diagnostic criteria and elaborate standard treatment protocol in JDM patients.
References
-
Feldman BM, Rider LG, Reed AM, Pachman LM (2008) Juvenile dermatomyositis and other idiopathic inflammatory myopathies of childhood. Lancet 371: 2201-2212.
-
Mendez EP, Lipton R, Ramsey-Goldman R, Roettcher P, Bowyer S, et al. (2003) "US incidence of juvenile dermatomyositis, 1995-1998: results from the National Institute of Arthritis and Musculoskeletal and Skin Diseases Registry". Arthritis Rheum 49: 300-305.
-
Kumar S, Pile K, Danda D (2013) Juvenile dermatomyositis management: moving but in need of a push. Int J Rheum Dis 16: 497-498.
-
Rider LG, Lindsley CB, Cassidy JT (2011) "Juvenile Dermatomyositis". In: Cassidy JT, Petty RE, Laxer RM, Lindsley CB, editors. "Textbook of Pediatric Rheumatology. 6th ed". Canada: Saunders Elsevier: 383.
-
Rider LG, Katz JD, Jones OY (2013) Developments in the classification and treatment of the juvenile idiopathic inflammatory myopathies. Rheum Dis Clin North Am 39: 877-904.
-
Bohan A, Peter JB (1975) Polymyositis and dermatomyositis". Parts 1 and 2. N Engl J Med 292: 344-347 and 292: 403-407
-
Findlay AR, Goyal NA, Mozaffar T (2015) An overview of polymyositis and dermatomyositis. Muscle Nerve .
-
Yu HH, Chang HM, Chiu CJ, Yang YH, Lee JH, et al. (2014) Detection of anti-p155/140, anti-p140, and antiendothelial cells autoantibodies in patients with juvenile dermatomyositis. J Microbiol Immunol Infect .
-
Bursac DS, Sazdanic-Velikic DS, Tepavac AP, Secen NM (2014) Paraneoplastic dermatomyositis associated with adenocarcinoma of the lung. J Cancer Res Ther 10: 730-732.
-
Fujita J, Tokuda M, Bandoh S, Yang Y, Fukunaga Y, et al. (2001) Primary lung cancer associated with polymyositis/dermatomyositis, with a review of the literature. Rheumatol Int 20: 81-84.
-
Marie I (2012) Morbidity and mortality in adult polymyositis and dermatomyositis. Curr Rheumatol Rep 14: 275-285.
-
Martin N, Krol P, Smith S, Beard L, Pilkington CA, et al. (2012) "Comparison of children with onset of juvenile dermatomyositis symptoms before or after their fifth birthday in a UK and Ireland juvenile dermatomyositis cohort study". Arthritis Care Res (Hoboken) 64: 1665-1672.
-
Sun C, Lee JH, Yang YH, Yu HH, Wang LC, et al. (2015) Juvenile dermatomyositis: a 20-year retrospective analysis of treatment and clinical outcomes. Pediatr Neonatol 56: 31-39.
-
Sanyal S, Atwal SS, Mondal D, Garga UC (2014) Radiographic patterns of soft tissue calcinosis in juvenile dermatomyositis and its clinical implications. J Clin Diagn Res 8: RD08-11.
-
Presley BC, Bush JS, Watson SC (2012) Dermatomyositis with extensive calcification in an adult. West J Emerg Med 13: 136-138.
-
Meher BK, Mishra P, Sivaraj P, Padhan P (2014) Severe calcinosis cutis with cutaneous ulceration in juvenile dermatomyositis. Indian Pediatr 51: 925-927.
-
Ishida T, Ohashi M, Matsumoto Y, Morikawa J, Sasaki R (1993) Connection of atopic disease in Japanese patients with juvenile dermatomyositis based on serum IgE levels. Clin Rheumatol 12: 41-48.
-
Habers GE, Van Brussel M, Bhansing KJ, Hoppenreijs EP, Janssen AJ, et al. (2014) Quantitative muscle ultrasonography in the follow-up of juvenile dermatomyositis. Muscle Nerve .
-
Huber AM, Robinson AB, Reed AM, Abramson L, Bout-Tabaku S, et al. (2012) "Consensus treatments for moderate juvenile dermatomyositis: beyond the first two months. Results of the second Childhood Arthritis and Rheumatology Research Alliance consensus conference". Arthritis Care Res (Hoboken) 64: 546-553.
-
Troyanov Y, Targoff IN, Payette MP, Raynauld JP, Chartier S, et al. (2014) "Redefining dermatomyositis: a description of new diagnostic criteria that differentiate pure dermatomyositis from overlap myositis with dermatomyositis features". Medicine (Baltimore) 93: 318-332.
