

Journal of Hypertension and Management
Preserved Left Ventricular Performance in Spontaneously Hypertensive Rats Following Preload and Afterload Challenges
Ming Fan, Mahmoud M. El-Mas, and Abdel A. Abdel-Rahman*
Department of Pharmacology, East Carolina University, USA
*Corresponding author:
Abdel A. Abdel-Rahman, PhD, Department of Pharmacology and Toxicology, School of Medicine, East Carolina University, Greenville, NC 27834, USA, Tel: 252-744-3470, Fax: 252-744-3203, E-mail: abdelrahmana@ecu.edu
J Hypertens Manag, JHM-1-002, (Volume 1, Issue 1), Original Research; ISSN: 2474-3690
Received: August 01, 2015 | Accepted: August 27, 2015 | Published: August 29, 2015
Citation: Fan M, El-Mas MM, Abdel-Rahman AA (2015) Preserved Left Ventricular Performance in Spontaneously Hypertensive Rats Following Preload and Afterload Challenges. J Hypertens Manag 1:002. 10.23937/2474-3690/1510002
Copyright: © 2015 Fan M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Hypertension-induced left ventricular (LV) hypertrophy is an independent risk factor for heart failure. To understand the impact of LV hypertrophy on cardiac hemodynamic before heart failure development, we studied LV performance and nitric oxide (NO) and reactive oxygen species (ROS) signaling in 6-month-old SHRs and age-matched Wistar-Kyoto rats (WKY) subjected to preload and afterload challenges. SHRs exhibited higher LV weight, LV developed pressure (LVDP), LV end-systolic pressure (LVESP), rate of rise (LV dp/dtmax) and fall (LV dp/dtmin) in LV pressure. Further, LV nNOS, but not eNOS, expression and nitrite/nitrate (NOx) were higher in SHRs than WKYs. Acute volume overload (dextrose infusion) caused smaller reductions in LV dp/dtmax and LVDP and greater increases in LVEDP and isovolumic relaxation constant (Tau, Τ) in SHRs, compared with WKYs, suggesting a better preserved systolic and depressed diastolic functions in SHRs. SHRs exhibited improved LV performance during pressure overload (phenylephrine infusion) as reflected by the greater increases in LVDP, LVESP, LV dp/dtmin, and LV dp/dtmax. LV ROS was increased by overload challenges in WKYs but not in SHRs. Overall, the blunted increase in LV ROS may account for the better-preserved ventricular performance during volume or pressure overload in SHRs despite evidence of early LV hypertrophy.
Keywords
Spontaneously hypertensive rats, Wistar Kyoto rats, Left ventricular hypertrophy, Left ventricular function, Nitric oxide synthase, Reactive oxygen species
Introduction
Chronic LV pressure overload, e.g. in hypertension, leads to cardiac hypertrophy and ultimately heart failure [1]. However, it is not clear if cardiac performance is compromised during early stages of LV hypertrophy before the development of heart failure. Studies in animals with LV hypertrophy revealed conflicting results due to differences in the magnitude, duration, and method of induction of cardiac hypertrophy as well as in the ventricular chamber involved [2-4]. The SHR is a well-established rat model of genetic hypertension, which resembles essential hypertension in humans [5]. LV hypertrophy in SHRs starts as early as 3 months [6] although the ejection fraction index-afterload relation could remain normal up to 18 months when heart failure starts to develop [7]. Higher [8] or lower [9] cardiac outputs have been reported in young SHR (3-4 months) compared with WKY rats. Moreover, Kobayashi et al. [4] showed increased contractility in myocytes isolated from 12-month-old SHRs.
Cardiac NO modulates cardiac functions both under physiological as well as pathophysiological conditions such as hypertension and heart failure [10,11]. eNOS, the dominant NOS isoform in vascular endothelium and cardiac myocytes, is an important regulator of adrenergic and muscarinic receptor signaling [12,13]. Moreover, eNOS is a prominent source of endothelial ROS in hypertension [14]. Alternatively, nNOS is detected in sarcoplasmic reticulum and cardiac conduction tissue and is involved in the modulation of the force and rhythm of myocardial contraction [15]. Despite increased NOS expression and NO generation in SHRs [16], findings on the role of NO in the cardiac contractile function, systemic hypertension, and left ventricular hypertrophy remain contradictory. For instance, NO may decrease [17], increase [18], or have no effect [19] on inotropic activity. These inconsistencies may relate to the use of different methods for induction of ventricular hypertrophy and whether anesthesia was employed. While at 6-months of age, the SHR represents an animal model for human hypertension with evidence of LV hypertrophy [20], the impact of preload and afterload challenges on cardiac performance in this animal model is not known.
In the current study, we tested the hypothesis that early LV hypertrophy in hypertensive rats alters LV performance in response to overload challenges. Accordingly, integrative cardiovascular studies were undertaken to evaluate LV function in 6-month-old conscious SHRs and age-matched WKY rats in response to volume or pressure overload challenges, which greatly impact LV performance [21]. The integrative studies were complemented with molecular studies to delineate the role of LV NOS expression/activity, and ROS level in LV performance at baseline and following pressure or volume overload in SHRs and in their controls. The data reported in this communication have been previously presented in the "Experimental Biology" meeting (EB2013) and published in an abstract form [22].
Materials and Methods
Age-matched (22-24 weeks) male SHR and WKY rats were obtained from Charles River (Raleigh, NC). Rats were acclimatized to laboratory conditions (23°C, 12h/12h light/dark, 50% humidity, ad libitum access to standard laboratory chow and water) for two weeks prior to experimentation. All experiments were approved by the institutional animal care and use committee and carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health.
Intravascular and intra ventricular cannulation
Vascular catheterization was conducted as in our previous studies [23-25]. Briefly, rats were anesthetized with i.p. injection of a mixture of ketamine (90 mg/kg) and xylazine (10 mg/kg). For intravascular cannulation, catheters, each consisting of 5-cm polyethylene-10 tubing bonded to 15-cm polyethylene-50 tubing, were placed into the abdominal aorta and vena cava via the left femoral vessels for measurement of arterial pressure and intravenous infusions, respectively. For the LV function measurement, polyethylene-50 tubing was inserted into the left ventricle through the carotid artery. Catheters were tunneled subcutaneously and exteriorized at the back of the neck between the scapulae. Catheters were filled with heparinized saline (100 U/ml) and plugged by stainless steel pins. Incisions were closed by surgical clips and swabbed with povidone-iodine solution. Each rat received intramuscular injections of the analgesic buprenorphine hydrochloride (Buprenex, 30 μg/kg) and penicillin G benzathine and penicillin G procaine in an aqueous suspension (Durapen, 100,000 U/kg). The catheter patency was maintained by daily heparin flushes. Two days later, hemodynamic measurements were made in conscious unrestrained rats.
Hemodynamic measurements and experimental groups
A total of 18 male SHRs and 18 age-matched WKY rats were divided into 3 groups for each strain (n = 6 each). Two groups (one SHR and one WKY) were used as controls for the measurement of basal LV NOx and ROS. The other four groups were used for the volume and pressure overload experiments. The arterial and left ventricular catheters were connected to pressure transducers. The data collected by these transducers are comparable to those produced by the Millar catheter [26].The pressure transducers were attached through MLAC11 Grass adapter cable to a computerized data acquisition system with LabChart-7 pro software (Power Lab, AD Instruments, Inc., Colorado, USA). The MAP, HR, LV dtmax, LV dtmin, LVESP, LVEDP, and LVDP were monitored. The isovolumic relaxation constant (Tau, Τ), which represents the exponential decay of the ventricular pressure during isovolumic relaxation, was also computed. At least 30 min stabilization period was allowed at the beginning of the experiment. After stabilization, baseline hemodynamic measurements were recorded.
Volume overload
To assess the effect of volume overload on LV performance, 5% dextrose was infused intravenously at low and high rates of 0.01 and0.03 ml/100g BW/s for 30s each based on a reported protocol [27]. Hemodynamic responses were recorded for 5 min after low-rate volume and for 50 min after the high-rate volume overload challenge.
Pressure overload
Phenylephrine was infused at a rate of 8 μg/kg/min to produce a steady-state elevation in blood pressure as described in reported studies including ours [28,29]. The selection of the phenylephrine dose regimen was also based on preliminary studies in which similar and sustained elevations in blood pressure were observed in the two rat strains. This was important to avoid the impact of discrepancies in the blood pressure response to phenylephrine on the measured LV functions. Hemodynamic monitoring continued for 120 min. Hemodynamic changes was analyzed using the PowerLab® data acquisition system (Power Lab, AD Instruments, Colorado Springs, CO).
Measurement of ventricular weights
At the conclusion of the experiment, euthanasia was induced with overdose of ketamine and xylazine mixture. The heart was excised immediately and atria and great vessels were removed. The ventricles were blotted free of blood. The left and right ventricles were separated and weighed. The ratio of the weight of the LV or RV to the body weight (mg/100 g) was computed.
Western blots
For the determination of myocardial eNOS and nNOS, the left ventricles from both strains of rats were homogenized on ice in a homogenization buffer [20 mM Tris (pH 7.5), 150 mM NaCL, 1 mM EGTA, 1 mM EDTA, 1% Triton X-100, 2.5 mM Sodium Pyrophosphate, 1 mM β-Glyceralphosphate, 1 mM Na3VO4, 1 mM Phenylmethylsulfonyl fluoride, 1 μg/ml leupetin, 1 tablet/20 ml complete protease inhibitor cocktail (Roche, Indianapolis, IN). After centrifugation (12,000 g for 10 min), protein in the supernatant was quantified (Bio-Rad protein assay system). Protein extracts (50 μg per lane) were run on a 4-12% SDS-PAGE gel (Invitrogen, CA) and electroblotted to nitrocellulose membranes. Blots were blocked for 120 min at room temperature in Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE). They were then incubated overnight at 4°C in Odyssey blocking buffer with mouse antibodies to eNOS 1:3000, nNOS 1:250, iNOS 1:5000 (BD Transduction Laboratories™, US) and rabbit antibody to GAPDH 1:2000 (Cell Signaling Technology, INC. Danvers, Massachusetts). After 3 washes with PBS-T buffer, the blots were incubated for 50 min at room temperature with a goat anti-mouse secondary antibody IRDye® 800CW or a goat anti-rabbit secondary antibody IRDye® 680 (LI-COR Biosciences, Lincoln, NE). After 3 washes with PBS-T buffer, the blots were detected by Odyssey Infrared Scanning System. The integrated intensity of the NOS protein bands was quantified by the Odyssey Infrared Imaging Software and expressed as a ratio to the GAPDH band intensity [30,31].
Measurement of LV nitrite/nitrate (NOx)
The ventricular NOx content was measured in triplicates using a fluorometric assay kit in accordance with the manufacturer's instructions (Cayman Chemical Company, Ann Arbor, MI) and as in our previous studies [23,32]. Briefly, 10 μl of enzyme cofactor and 10 μl of nitrate reductase were added to 80 μl sample mixture, incubated at room temperature for 3 hr. Fifty μl of Griess Regent R1 and R2 (Cayman Chemical Company, Ann Arbor, MI) was then added and the absorbance was read at 540 nm.
Measurement of LV ROS
The method described by LeBel et al. [33] was used to measure total ROS/RNS including superoxide, hydroxyl radical, and hydrogen peroxide. Briefly, 2, 7-dichlorofluorescin diacetate (DCFH-DA, Molecular Probes, Inc., Eugene, OR) was dissolved in absolute ethanol at a concentration of 12.5 mM and kept at -80°C and protected from light. It was diluted with 50 mM phosphate buffer (pH 7.4) to a final concentration of 125 μM. DCFH-DA was added to LV homogenate in a 96-well plate to achieve a final concentration of 25 μM. The plates were incubated at 37°C for 30 min in the presence of esterase (0.3 U) to ascertain a steady-level of 2,7-dichlorofluorescein. The change in fluorescence intensity was monitored at two time points (0 and 30 min) using a microplate fluorescence reader (Bio-TEK Instruments, Inc., Winooski, VT) at excitation 485 nm/emission 530 nm. ROS/RNS status was calculated as fluorescence per milligram of protein and per min. Samples were measured in triplicates.
Drugs
Xylazine, ketamine, Durapen (Penicillin G benzathine and penicillin G procaine), and buprenex (buprenorphine hydrochloride) were provided by East Carolina University, Department of Comparative Medicine (Webster Veterinary Supplies Vortech Pharmaceutical Ltd., Dearborn, MI). Dextrose and phenylephrine hydrochloride were purchased from Sigma Chemical (St. Louis, MO), and povidone-iodine solution from Norton (Rockford, IL).
Data analysis and Statistics
Values are presented as means ± S.D. Normal distribution was checked using column statistics (modified Kolmogorov-Smirnov test, GraphPad Prism, software release 5.0). Mean arterial pressure (MAP) was calculated as diastolic pressure + one-third pulse pressure (systolic - diastolic pressures). Baseline body weight, ventricular weight and cardiovascular variables were pooled from all SHR and WKY rats (n = 18 each). Unpaired Student t test was used for simple comparisons. The repeated measures analysis of variance (ANOVA) followed by a Newman-Keuls post-hoc test was used for multiple comparisons. Statistical significance was defined as p < 0.05.
Results
Ventricular weights and cardiovascular variables
Table 1 illustrates the body and ventricular weights and basal cardiovascular parameters for SHR and WKY rats. The absolute LV weight and the LV/body weight ratios were significantly (P < 0.05) higher in SHRs (Table 1). The RV weight and the RV/body weight ratios were similar in the two strains of rats (Table 1). As expected, MAP was significantly (P < 0.05) higher in SHR than in WKY rats (Table 1). Measurements of LV performance revealed significantly (P < 0.05) higher LVESP, LVDP, LV dtmax, and LV dtmin in SHRs (Table 1). There was no difference in LVEDP or isovolumic relaxation constant (Τ) between the two strains (Table 1).The LV NO metabolites (Table 1) and nNOS, but not eNOS, protein expression (Figure 1) were significantly (P < 0.05) higher in SHRs than in WKY rats.
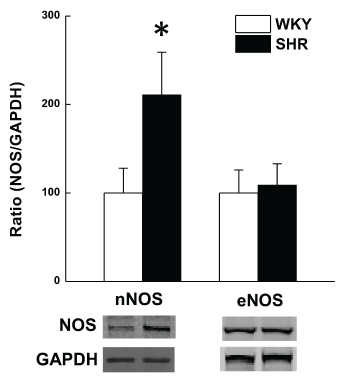
.
Figure 1: The protein expression of nNOS and eNOS in LV of SHR and WKY rats under basal conditions. Values are means ± S.D. of 6 observations. *p < 0.05 compared with WKY values.
View Figure 1
![]()
Table 1: Body weights and left (LV) and right (RV) ventricular weights and their ratios in WKYs and SHRs. The baseline values of different cardiovascular variables and LV NOx contents in the two rat strains are also shown.
View Table 1
Strain-dependent effects of volume or pressure overload on LV performance
To determine the impact of volume or pressure overload on cardiac function in SHR vs. WKY rats, the changes in hemodynamic parameters were compared after subjecting the animals to low- and high-rate volume or pressure overload challenge. The data were presented as percentages of basal values since there were marked differences in basal values between the two strains. In both strains of rats, low- and high-rate of 5% dextrose infusion (0.01 and 0.03 ml/100 g BW/s, respectively) for 30 seconds elicited volume-dependent reductions in LV dtmax, LV dtmin, LVDP, and MAP (Figure 2A, Figure B, Figure C and Figure F), and increases in LVEDP (Figure 2E). These hemodynamic responses started during the dextrose infusion, reached steady state within 5 min, and dissipated after approximately 30 min (Fig. 2A-F). Nonetheless, the reductions in cardiac indices were less pronounced in SHRs, particularly, the significantly (P < 0.05) smaller reductions in LV dtmax (Figure 2B). By contrast, the increases in LVEDP were substantially (P < 0.05) more pronounced in SHRs (Figure 2E) indicating better preserved LV contractile function during volume load in SHRs. On the other hand, the ratio of LV dtmax to LV dtmin was significantly higher in SHR compared with WKY rats (1.37 ± 0.07 vs. 1.04 ± 0.12, p < 0.05) indicating a negative lusitropic pattern in hypertrophic SHR hearts. No significant changes in HR were observed in either rat strain (data not shown). The isovolumic relaxation constant (Τ) was also higher in SHRs compared with WKY rats under low-rate volume overload challenge (13.46 ± 2.75% vs. 7.41 ± 1.01% increase). The Τ increased in both strains of rats with high-rate volume overload challenge but no significant differences were observed.
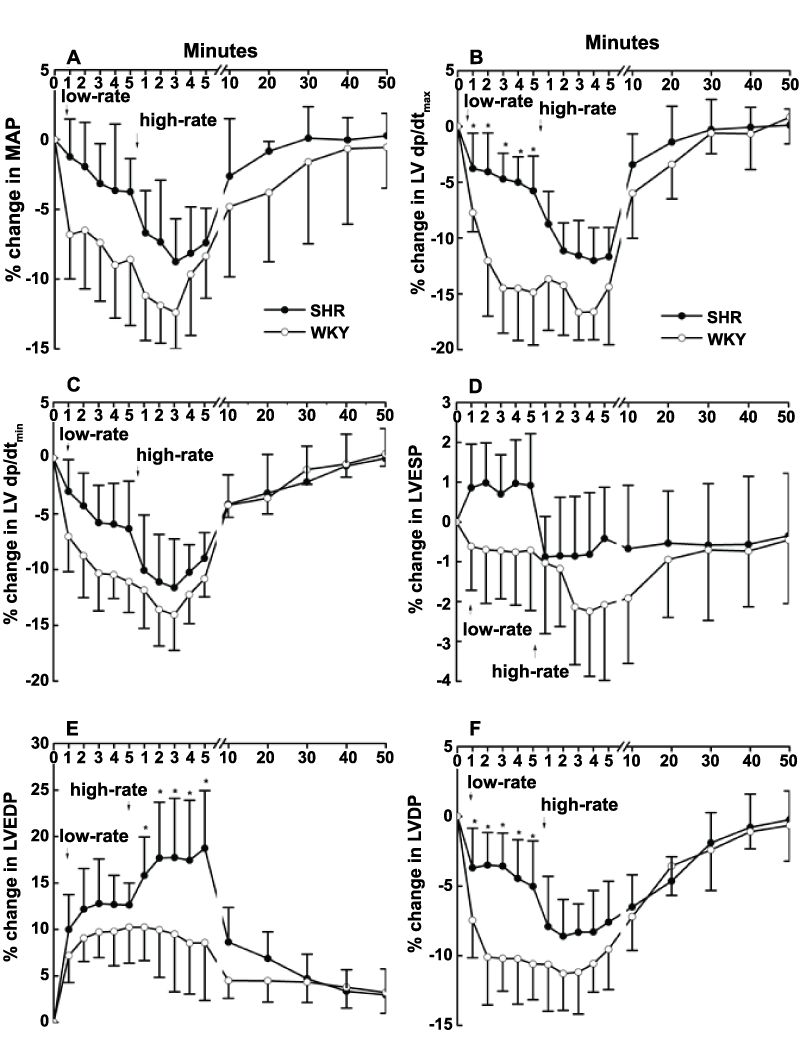
.
Figure 2: Percentage changes in hemodynamics after 30-second infusion of 5% dextrose at rates of 0.01ml (low-rate) and 0.03 ml (high-rate)/100g BW/s in SHR and WKY rats (see materials and methods for details). Values are means ± S.D. of 6 observations. *p < 0.05 compared with WKY values.
View Figure 2
Pressure overload caused by phenylephrine infusion (8 μg/kg/min) elicited similar steady-state elevations in MAP in the two strains of rats when presented as percentages of baseline pressure values (Figure 3A). Similarly, the absolute increases in MAP caused by phenylephrine infusion in SHRs and WKY rats were not statistically different (e.g. 21 ± 6 vs. 17 ± 8 mmHg after 100 min of phenylephrine infusion). However, under similar pressure overload conditions (Figure 3A), SHRs exhibited significantly (P < 0.05) greater increases in LV dtmax, LV dtmin, LVESP and LVDP compared with WKY rats (Figure 3B, Figure C, Figure D and Figure F). Unlike the volume challenge, no differences in the ratio of LV dtmax to LV dtmin or isovolumic relaxation constant were observed between the two strains (data not shown). HR was slightly decreased during phenylephrine infusion in strain-independent manner (SHRs, -4.7 ± 3.8%; WKYs, -2.6 ± 0.8%, P > 0.05).
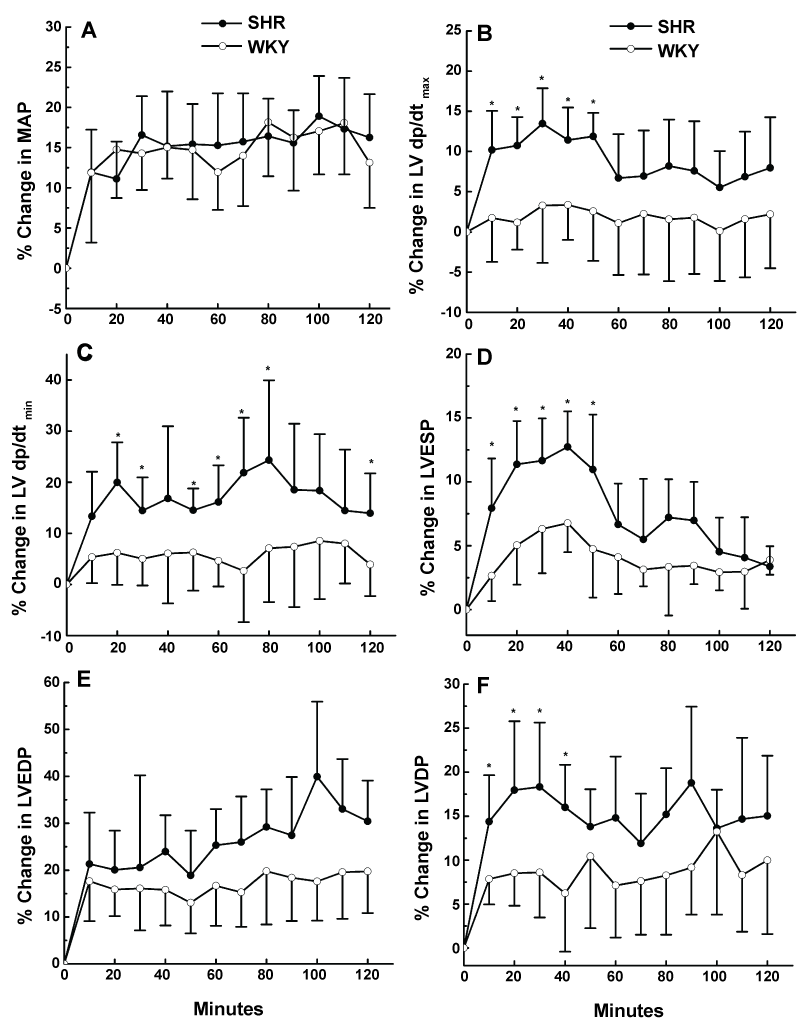
.
Figure 3: Percentage changes in hemodynamics during the infusion of phenylephrine at a rate of 8 μl/kg/min in SHR and WKY rats (see materials and methods for details). Values are means ± S.D. of 6 observations. *p < 0.05 compared with WKY values.
View Figure 3
Volume or pressure overload increases LV ROS level in WKY rats
Basal ROS levels were slightly but not significantly higher in LV tissues of SHRs compared with WKY rats (Figure 4). On the other hand, the volume or pressure overload challenge significantly (P < 0.05) elevated LV ROS in WKY rats in contrast to no effect in SHRs (Figure 4).
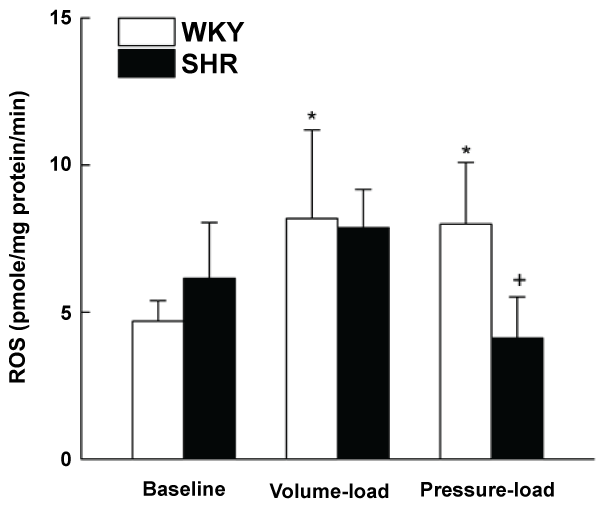
.
Figures 4: Levels of reactive oxygen species (ROS, pmole/mg protein/min) in LV of SHR and WKY rats under basal conditions and following overload challenges (volume or pressure). Values are means ± S.D. of 6 observations. *p < 0.05 compared with basal values in WKY rats, +p < 0.05 compared with pressure-load values in WKY rats.
View Figure 4
Discussion
In the present study we investigated the effects of preload and afterload challenges on LV performance in the presence of LV hypertrophy in conscious unrestrained 6-month-old SHRs as compared to age-matched WKY rats. The main findings of this study were: 1) at baseline conditions, SHRs exhibited LV hypertrophy accompanied by increases in LV contractility and unaltered LV diastolic function; 2) an adaptive upregulation of LV nNOS exists in SHRs; 3) preload challenge, by rapid elevation of volume load of LV, caused less reductions in LV dtmax and LVDP, and prolongedthe isovolumic relaxation constant, in SHRs; 4) SHRs manifested enhanced LV contractility in response to the pressure load challenge, compared with WKY rats; and 5) the differences between in LV responses of SHR and WKY rats to overload challenges (volume or pressure) can be attributed, at least partly, to inter-strain differences in LV ROS generation. Collectively, the data suggest that despite LV hypertrophy, the SHR exhibits better LV function at basal level, and following interventions that cause deficits in LV function when compared to WKY (control) rats.
LV mass (LV/BW ratio) was significantly higher in SHRs compared with WKY rats, which is consistent with LV hypertrophy in hypertensive rats as reported elsewhere [34]. However, contrary to what we expected, SHRs exhibited significantly higher LV dtmax, LV dtmin, LVESP and LVDP indicating increased basal LV contractile function. Notably, LV performance of hypertrophied hearts in both experimental animals and in patients has been reported as normal [35], enhanced [36] or depressed [37]. In support of our results, McCrossan et al. [38] demonstrated that SHRs, similar in age to those in our study, exhibited concentric hypertrophy with larger volume of cardiomyocytes and significantly higher myocardial contractility, compared to age-matched WKY rats. Cingolani et al. [34] found that LV dtmax and LV maximum dp/dt-end-diastolic volume relation were higher in 10-month old SHRs. Nonetheless, unlike our current study in which SHRs showed normal LV diastolic function at baseline conditions, the SHRs exhibited end-diastolic stiffness and diastolic dysfunction in other studies [34]. This discrepancy may be due to the use of older SHRs (10 months) in the latter study [34], compared with the 6 month-old SHRs used in our study.
The role of endogenous NO production in the regulation of ventricular performance remains unclear. Ashley et al. [39] demonstrated that cardiomyocyte contraction increased when nNOS activity was suppressed by genetic disruption or pharmacological inhibition. Enhanced LV systolic function and elevated LV ejection fraction were seen in nNOS-/-, compared with the wild type [40]. These findings indicated negative regulation of myocardial contraction by nNOS. In our study, nNOS, but not eNOS, expression was significantly higher in LV of SHRs compared with WKY rats (Figure 1). Notably, the higher NOx level in the same LV tissues of SHR (Table 1), which is consistent with reported findings [41], supports the involvement of the upregulated nNOS in the higher LV NO in SHRs. The upregulation of nNOS in LV of SHR may be a regulatory mechanism to maintain energetic homeostasis in performance-enhanced hearts of SHRs. Unlike nNOS, LVeNOS expression was similar in SHRs and WKY rats (Figure 1). This finding may contrast with reported NOS involvement in ventricular performance. First, in eNOS knockout murine model, chronic pressure-overload results in myocyte hypertrophy, LV fibrosis, and impaired systolic contractility and diastolic relaxation [42]. Second, eNOS over expression in the mouse myocardium enhances cardiac NO generation and reduced LV contractility [43]. Because endogenous NO exerted a positive inotropic effect in the wild type animal, a dual myocardial regulatory activity for eNOS on myocardial contractility has been suggested [44]. It is likely that these different findings relate to species, strain and age differences and whether anesthesia was used or not. More studies are needed to address these issues.
With the exception of LVEDP, all indices of systolic and diastolic performance were negatively impacted by the rapid infusion of 5% dextrose (0.01 and 0.03 ml/100g BW/s) in the two rats strains (Figure 2). This finding is consistent with reported studies, which suggested a role for endocardial ischemia and reduced transient Ca2+ currents in ventricular dysfunction induced by volume overload [44]. Interestingly, our data revealed remarkable strain-dependent differences in the LV response to the volume overload challenge. Most notably, significantly smaller reductions in LV dtmax and LVDP were observed in SHRs compared with WKY rats. This finding suggests that at the age of the rats used in the present study (6 months), the systolic function of SHRs is better protected against the detrimental effects of volume overload.
Given the substantially higher LV dtmax to LV dtmin ratio in SHR, than in WKY, rats, it is likely that a more negative lusitropic pattern existed in the hypertrophic heart of SHRs. Indeed, the prolongation of the isovolumic relaxation constant (Τ), which reflects cardiac diastolic function [45], in SHRs compared with WKY rats, might infer a delayed LV relaxation in response to volume overload in SHRs. Such reductions in LV compliance may be accounted for by the existing hypertrophied LV. Together; the present study is the first to demonstrate the contrasting LV consequences of the volume overload challenge, enhanced systolic and worsened diastolic performance in SHRs.
The LV performance is also influenced by afterload. Aortic constriction and phenylephrine infusion are effective maneuvers to test the influence of pressure overload on LV function [21,29,46]. In the current study, we examined the impact of sustained steady-state elevations in afterload, induced by phenylephrine infusion, on LV performance. In order to permit appropriate inter-strain comparisons, we selected an infusion regimen of phenylephrine that caused similar (~ 15%) and sustained elevations in arterial pressure throughout the 2-hr observation period (Figure 3A). While such intervention exerted little effect on LV performance in WKY rats, SHRs exhibited significant increases in LV dtmax, LV dtmin, LVESP and LVDP for approximately 50 min (Figure 3). These exciting new findings suggest that the adaptive LV hypertrophy, due probably to chronic hypertension, might enable the SHR to preserve LV performance following sudden increase in the afterload.
Although growing evidence implicates oxidative stress in the development of LV hypertrophy [47,48], it is not known if oxidative stress is the cause or consequence of ventricular hypertrophy [49].We show that myocardial ROS level, a well accepted measure of oxidative stress [14,33], was similar in SHRs in presence of LV hypertrophy and in their controls (Figure 4). This finding suggests that LV hypertrophy in 6-month old SHRs is not dependent on oxidative stress. This finding is partly supported by the reported observations that while ventricular hypertrophy developed in relatively young SHRs (4-month-old), increased generation of ventricular ROS along with more deterioration in the hypertrophied LV performance became manifest in SHRs at the age of 11 months and older [49]. Equally important, the similar basal LV ROS levels in the two rat strains argue against a role for oxidative stress in the SHR-WKY differences in LV performance. Nonetheless, we show here that the LV overload challenges (volume or pressure) increased the ventricular ROS levels in WKY rats in contrast to no effect in SHRs. Therefore, it is plausible to suggest that the ability of the SHR LV to resist the induction of oxidative stress might explain, at least partly, the better (preserved) LV performance following volume or pressure challenges in SHRs compared with WKY rats. It is unlikely, however, that the deteriorated oxidative state in phenylephrine-treated WKY rats has contributed to the concomitant increases in blood pressure because similar blood pressure rises were observed in the two rat strains (Figure 3A). This view is supported by the study of Zhang et al. [50], which failed to establish any correlation between the hypertensive and LV oxidative response elicited by phenylephrine or other vasopressor agents, e.g. angiotensin II, in normotensive rats.
In conclusion, compared with age-matched WKY rats, 6-month old SHRs manifested LV hypertrophy along with enhanced LV contractility and normal diastolic function. An increase in nNOS expression in LV of SHRs may be a regulatory mechanism for enhanced LV contractility. The adaptive LV hypertrophy in SHRs permitted the sustainability of the increase in LV contractile performance in response to pressure-load challenge. The acute volume overload challenge improved the systolic performance and induced negative lusitropism in SHRs. The increased generation of LV ROS in WKY rats might contribute to the LV deficits caused by overload challenges while the ability of the SHR to resist the induction of LV ROS generation might explain the better preserved LV function under similar overload conditions in this model of genetic hypertension.
Acknowledgments
This work was supported by the National Institute on Alcohol Abuse and Alcoholism [2R01 AA07839-19]. The authors thank Ms. Kui Sun for her technical assistance.
Footnotes
Dr. Mahmoud. M. El-Mas is a Visiting Professor from the Department of Pharmacology, Faculty of Pharmacy, Alexandria University, Egypt (Email: mahelm@hotmail.com).
References
-
Luedde M, Katus HA, Frey N (2006) Novel molecular targets in the treatment of cardiac hypertrophy. Recent Pat Cardiovasc Drug Discov 1: 1-20.
-
Brower GL, Janicki JS (2001) Contribution of ventricular remodeling to pathogenesis of heart failure in rats. Am J Physiol Heart Circ Physiol 280: H674-H683.
-
Capasso JM, Palackal T, Olivetti G, Anversa P (1990) Left ventricular failure induced by long-term hypertension in rats. Circ Res 66: 1400-1412.
-
Kobayashi T, Hamada M, Okayama H, Shigematsu Y, Sumimoto T, et al. (1995) Contractile properties of left ventricular myocytes isolated from spontaneously hypertensive rats: effect of angiotensin II. J Hypertens 13: 1803-1807.
-
Boluyt MO, Bing OH, Lakatta EG (1995) The ageing spontaneously hypertensive rat as a model of the transition from stable compensated hypertrophy to heart failure. Eur Heart J 16 Suppl N: 19-30.
-
Hasenfuss G (1998) Animal models of human cardiovascular disease, heart failure and hypertrophy. Cardiovasc Res 39: 60-76.
-
Mirsky I, Pfeffer JM, Pfeffer MA, Braunwald E (1983) The contractile state as the major determinant in the evolution of left ventricular dysfunction in the spontaneously hypertensive rat. Circ Res 53: 767-778.
-
Weisser-Thomas J, Nguyen Q, Schuettel M, Thomas D, Dreiner U, et al. (2007) Age and hypertrophy related changes in contractile post-rest behavior and action potential properties in isolated rat myocytes. Age (Dordr) 29: 205-217.
-
El-Mas MM, Abdel-Rahman AA (1999) Acute hemodynamic effects of ethanol in conscious spontaneously hypertensive and normotensive rats. Alcohol Clin Exp Res 23: 285-292.
-
Kanai AJ, Mesaros S, Finkel MS, Oddis CV, Birder LA, et al. (1997) Beta-adrenergic regulation of constitutive nitric oxide synthase in cardiac myocytes. Am J Physiol 273: C1371-1377.
-
Casadei B, Sears CE (2003) Nitric-oxide-mediated regulation of cardiac contractility and stretch responses. Prog Biophys Mol Biol 82: 67-80.
-
Massion PB, Balligand JL (2003) Modulation of cardiac contraction, relaxation and rate by the endothelial nitric oxide synthase (eNOS): lessons from genetically modified mice. J Physiol 546: 63-75.
-
Champion HC, Georgakopoulos D, Takimoto E, Isoda T, Wang Y, et al. (2004) Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ Res 94: 657-663.
-
Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, et al. (2003) Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201-1209.
-
Kelly RA, Balligand JL, Smith TW (1996) Nitric oxide and cardiac function. Circ Res 79: 363-380.
-
Chou TC, Yen MH, Li CY, Ding YA (1998) Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 31: 643-648.
-
Flesch M, Kilter H, Cremers B, Lenz O, Südkamp M, et al. (1997) Acute effects of nitric oxide and cyclic GMP on human myocardial contractility. J Pharmacol Exp Ther 281: 1340-1349.
-
Cotton JM, Kearney MT, MacCarthy PA, Grocott-Mason RM, McClean DR, et al. (2001) Effects of nitric oxide synthase inhibition on Basalfunction and the force-frequency relationship in the normal and failing human heart in vivo. Circulation 104: 2318-2323.
-
Ungureanu-Longrois D, Bézie Y, Perret C, Laurent S (1997) Effects of exogenous and endogenous nitric oxide on the contractile function of cultured chick embryo ventricular myocytes. J Mol Cell Cardiol 29: 677-687.
-
Pfeffer JM, Pfeffer MA, Fishbein MC, Frohlich ED (1979) Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am J Physiol 237: H461-H468.
-
Friberg P (1985) Structural and functional adaptation in the rat myocardium and coronary vascular bed caused by changes in pressure and volume load. Acta Physiol Scand Suppl 540: 1-47.
-
Fan M, El-Mas MM, Abdel-rahman AA (2013) Preserved left ventricular performance in spontaneously hypertensive rats following preload and afterload challenges. FASEB J 27: 654.
-
El-Mas MM, Fan M, Abdel-Rahman AA (2009) Facilitation of myocardial PI3K/Akt/nNOS signaling contributes to ethanol-evoked hypotension in female rats. Alcohol Clin Exp Res 33: 1158-1168.
-
El-Mas MM, Abdel-Rahman AA (1997) Aortic barodenervation upregulates? 2-adrenoceptors in the nucleus tractus solitarius and rostral ventrolateral medulla: an autoradiographic study. Neuroscience 79: 581-590.
-
El-Mas MM, Abdel-Rahman AA (2014) Nongenomic effects of estrogen mediate the dose-related myocardial oxidative stress and dysfunction caused by acute ethanol in female rats. Am J Physiol Endocrinol Metab 306: E740-747.
-
Wang Q, Brunner HR, Burnier M (2004) Determination of cardiac contractility in awake unsedated mice with a fluid-filled catheter. Am J Physiol Heart Circ Physiol 286: H806-H814.
-
Huang BS, white RA, Ahmad M, Tan J, Jeng AY (2009) Central infusion of aldosterone synthase inhibitor attenuates left ventricular dysfunction and remodeling in rats after myocardial infarction. Cardiovasc Res 81: 574-581.
-
El-Mas MM, Zhang J, Abdel-Rahman AA (2006) Upregulation of vascular inducible nitric oxide synthase mediates the hypotensive effect of ethanol in conscious female rats. J Appl Physiol (1985) 100: 1011-1018.
-
Magga J, Marttila M, Mäntymaa P, Vuolteenaho O, Ruskoaho H (1994) Brain natriuretic peptide in plasma, atria, and ventricles of vasopressin- and phenylephrine-infused conscious rats. Endocrinology 134: 2505-2515.
-
Zhang J, El-Mas MM, Abdel-Rahman AA (2001) Imidazoline I (1) receptor-induced activation of phosphatidylcholine-specific phospholipase C elicits mitogen-activated protein kinase phosphorylation in PC12 cells. Eur J Pharmacol 415: 117-125.
-
El-Mas MM, El-Gowelli HM, Abd-Elrahman KS, Saad EI, Abdel-Galil AG, et al. (2011) Pioglitazone abrogates cyclosporine-evoked hypertension via rectifying abnormalities in vascular endothelial function. Biochem Pharmacol 81: 526-533.
-
El-Mas MM, El-Gowilly SM, Gohar EY, Ghazal AR (2008) Pharmacological characterization of cellular mechanisms of the renal vasodilatory effect of nicotine in rats. Eur J Pharmacol 588: 294-300.
-
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2',7'-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5: 227-231.
-
Cingolani OH, Yang XP, Cavasin MA, Carretero OA (2003) Increased systolic performance with diastolic dysfunction in adult spontaneously hypertensive rats. Hypertension 41: 249-254.
-
Sasayama S, Franklin D, Ross J Jr (1977) Hyperfunction with normal inotropic state of the hypertrophied left ventricle. Am J Physiol 232: H418-H425.
-
Malik AB, Abe T, O'kane H, Geha AS (1973) Cardiac function, coronary flow, and oxygen consumption in stable left ventricular hypertrophy. Am J Physiol 225: 186-191.
-
Kokubo M, Uemura A, Matsubara T, Murohara T (2005) Noninvasive evaluation of the time course of change in cardiac function in spontaneously hypertensive rats by echocardiography. Hypertens Res 28: 601-609.
-
McCrossan ZA, Billeter R, White E (2004) Transmural changes in size, contractile and electrical properties of SHR left ventricular myocytes during compensated hypertrophy. Cardiovasc Res 63: 283-292.
-
Ashley EA, Sears CE, Bryant SM, Watkins HC, Casadei B (2002) Cardiac nitric oxide synthase 1 regulates basal and beta-adrenergic contractility in murine ventricular myocytes. Circulation 105: 3011-3016.
-
Sears CE, Bryant SM, Ashley EA, Lygate CA, Rakovic S, et al. (2003) Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res 92: e52-e59.
-
Hayakawa H, Raij L (1997) The link among nitric oxide synthase activity, endothelial function, and aortic and ventricular hypertrophy in hypertension. Hypertension 29: 235-241.
-
Ichinose F, Bloch KD, Wu JC, Hataishi R, Aretz HT, et al. (2004) Pressure overload-induced LV hypertrophy and dysfunction in mice are exacerbated by congenital NOS3 deficiency. Am J Physiol Heart Circ Physiol 286: H1070-H1075.
-
Brunner F, Andrew P, Wolkart G, Zechner R, Mayer B (2001) Myocardial contractile function and heart rate in mice with myocyte-specific overexpression of endothelial nitric oxide synthase. Circulation 104: 3097-3102.
-
Holt W, Auffermann W, Wu ST, Parmley WW, Wikman-Coffelt J (1991) Mechanism for depressed cardiac function in left ventricular volume overload. Am Heart J 121: 531-537.
-
Matsubara H, Takaki M, Yasuhara S, Araki J, Suga H (1995) Logistic time constant of isovolumic relaxation pressure-time curve in the canine left ventricle. Better alternative to exponential time constant. Circulation 92: 2318-2326.
-
Liakopoulos OJ, Ho JK, Yezbick AB, Sanchez E, Singh V, et al. (2010) Right ventricular failure resulting from pressure overload: role of intra-aortic balloon counterpulsation and vasopressor therapy. J Surg Res 164: 58-66.
-
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115: 500-508.
-
Alter P, Rupp H, Rominger MB, Figiel JH, Renz H, et al. (2010) Association of hyperhomocysteinemia with left ventricular dilatation and mass in human heart. Clin Chem Lab Med 48: 555-560.
-
Alvarez MC, Caldiz C, Fantinelli JC, Garciarena CD, Console GM, et al. (2008) Is cardiac hypertrophy in spontaneously hypertensive rats the cause or the consequence of oxidative stress? Hypertens Res 31: 1465-1476.
-
Zhang GX, Kimura S, Nishiyama A, Shokoji T, Rahman M, et al. (2004) ROS during the acute phase of Ang II hypertension participates in cardiovascular MAPK activation but not vasoconstriction. Hypertension 43: 117-124.
