We present an unusual case of a 40-year-old adult man with Antithrombin III deficiency who initially presented with cerebral arterial thrombosis and subsequently developed cerebral venous thrombosis during treatment.
The subject was a 40-year-old man with a known past history of mild systemic hypertension, hyperlipidemia and ischemic heart disease. He woke up six hours prior to presentation and found that he was unable to move the left limbs. He was on Aspirin 100 mg once daily, Atorvastatin 10 mg once daily and Carvedilol 6.25 mg once daily. He had undergone coronary angioplasty fifteen years previously.
On examination he was conscious and alert with left facial weakness and dense left hemiplegia with power MRC grade 0/5 on the left side. Tendon reflexes were absent on the left side and left plantar was extensor. Sensation was normal on the left side. No hemianopia was evident clinically. NIH score was 10.
MRI brain done immediately showed patchy diffusion restriction in whole of the right middle cerebral artery (MCA) territory, however, only minimal insular involvement was seen on FLAIR sequence. Thus there was diffusion and FLAIR mismatch. MRA showed complete cut off at proximal right MCA consistent with large vessel occlusion (LVO) (Figure 1 and Figure 2).
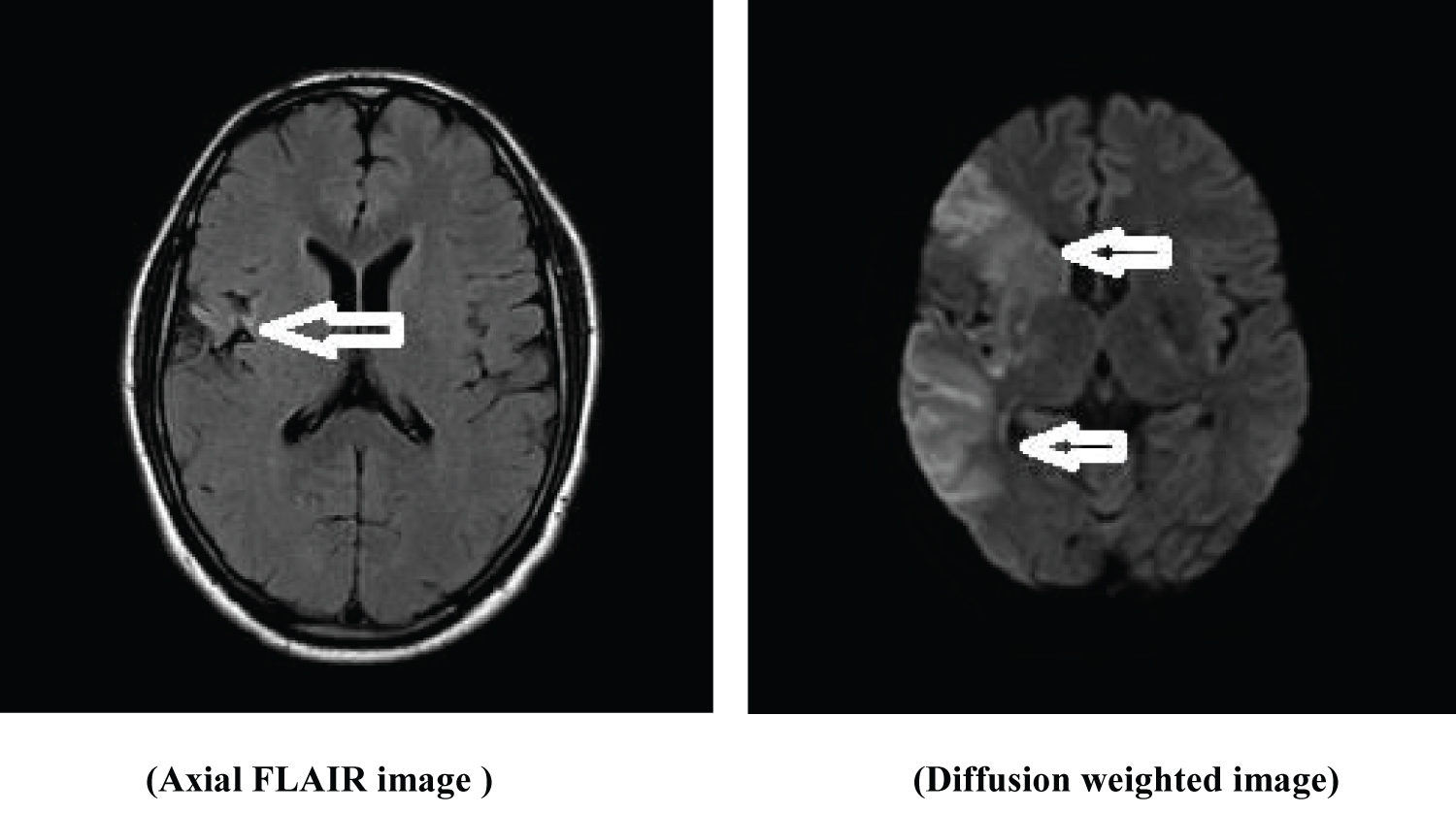 Figure 1: MRI images showing FLAIR-Diffusion mismatch.
View Figure Chart 1
Figure 1: MRI images showing FLAIR-Diffusion mismatch.
View Figure Chart 1
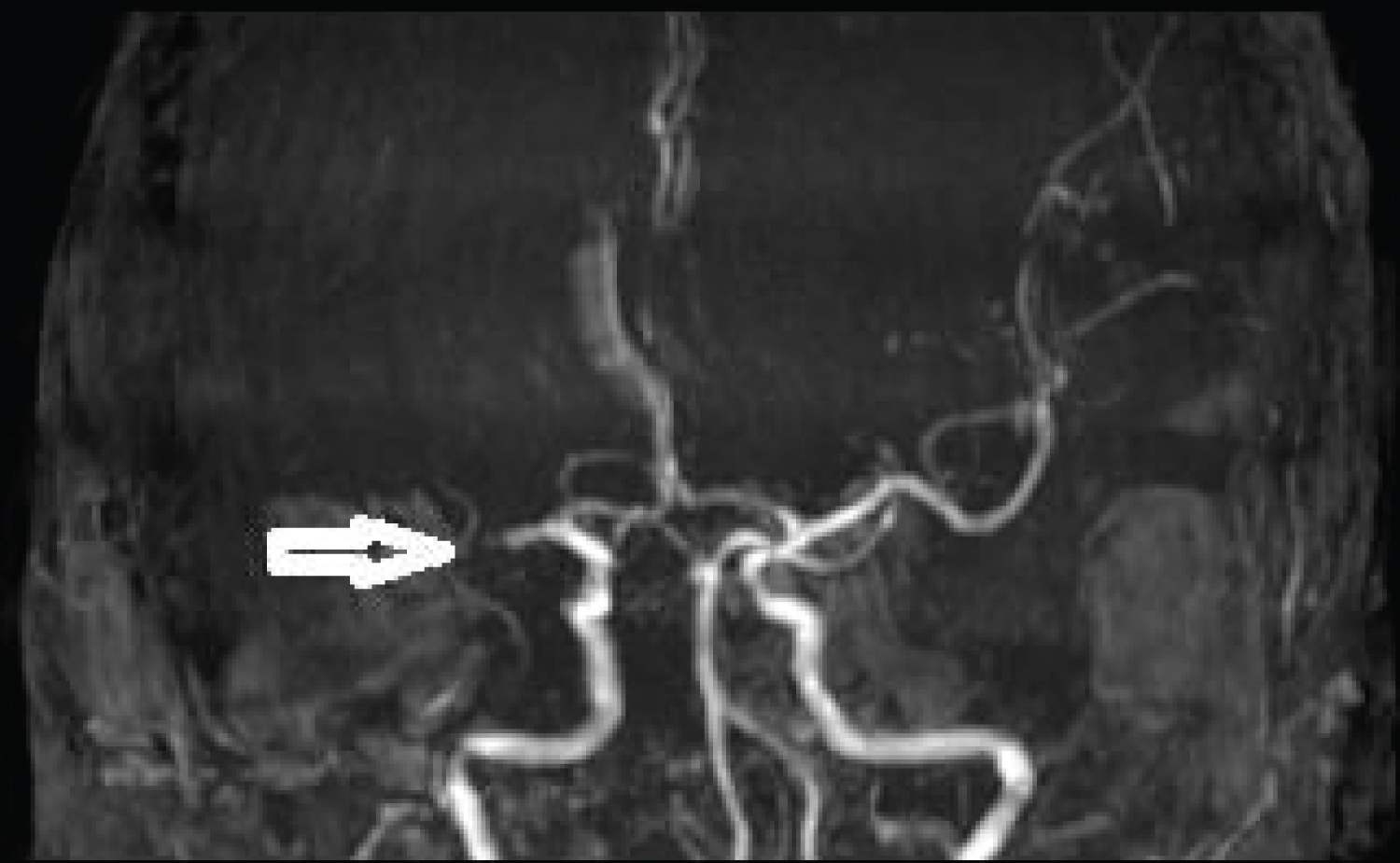 Figure 2: Non-contrast MRA showing complete cut off of right MCA. View Figure Chart 2
Figure 2: Non-contrast MRA showing complete cut off of right MCA. View Figure Chart 2
The subject was beyond the window period for intravenous thrombolysis, however in view of LVO and diffusion Flair mismatch, he was a candidate for mechanical thrombectomy. He underwent mechanical thrombectomy soon after and had complete reperfusion of the right MCA. He was noted to have hemorrhagic transformation of the right MCA infarct post mechanical thrombectomy on the CT scans of the head done hours after post thrombectomy as well as on day three post stroke. He was however continued on aspirin 100 mg once daily. He was started on mannitol 20% for brain edema. Clinically he continued to improve. On day six post stroke on examination he had only mild left facial weakness with power in the left limbs improved to MRC grade 4/5 in the left upper limb and 5/5 in the left lower limb. He was walking independently.
MRI brain on day six post stroke showed a wedge shaped hypodensity in the right frontal and parietal region consistent with infarct with edema and mass effect with minimal midline shift 3 mm. Hemorrhagic change was seen in the area of the basal ganglia (Figure 3 and Figure 4).
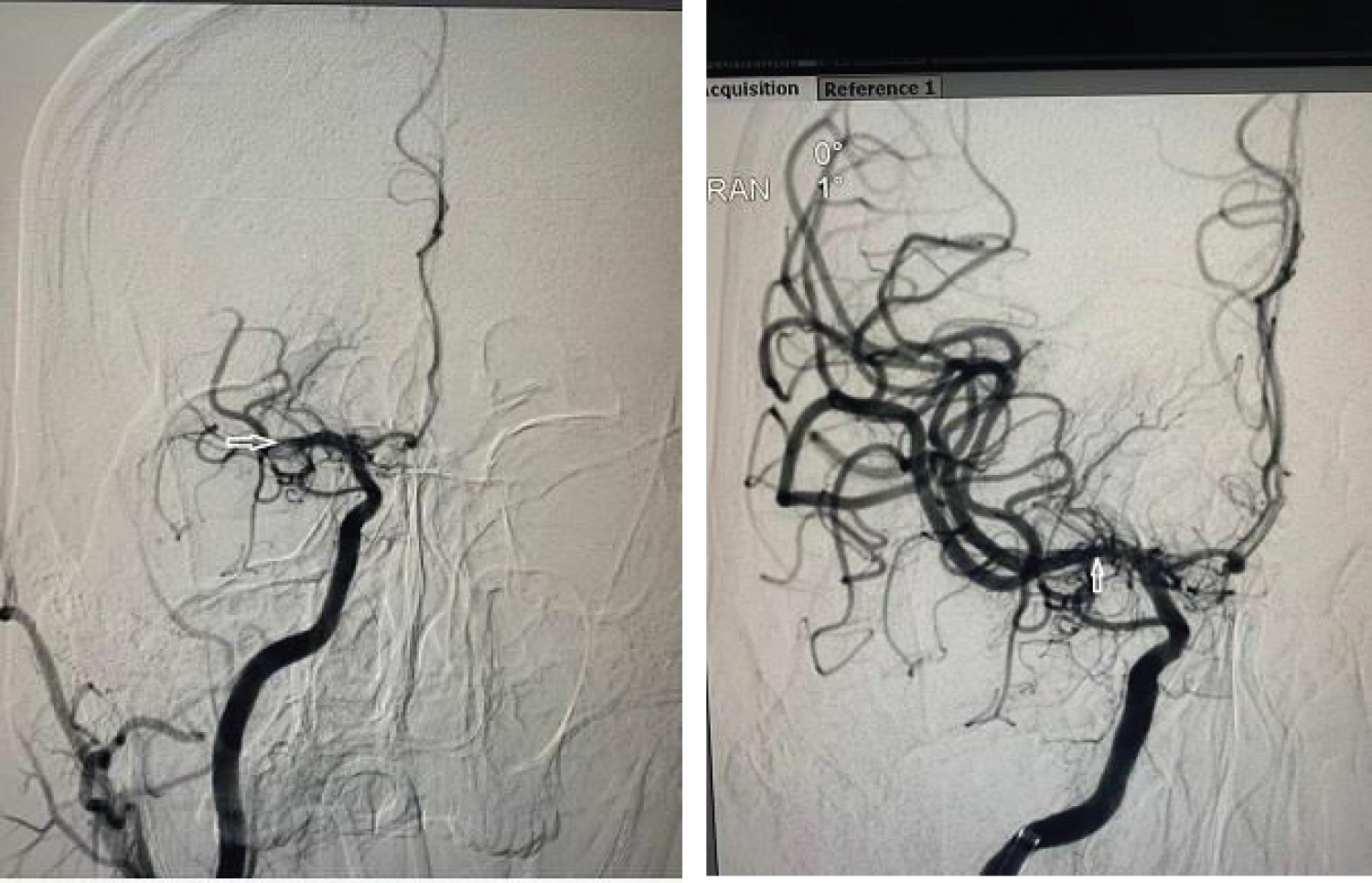 Figure 3: Pre and post mechanical thrombectomy DSA shows occluded and opened up Right MCA respectively.
View Figure Chart 3
Figure 3: Pre and post mechanical thrombectomy DSA shows occluded and opened up Right MCA respectively.
View Figure Chart 3
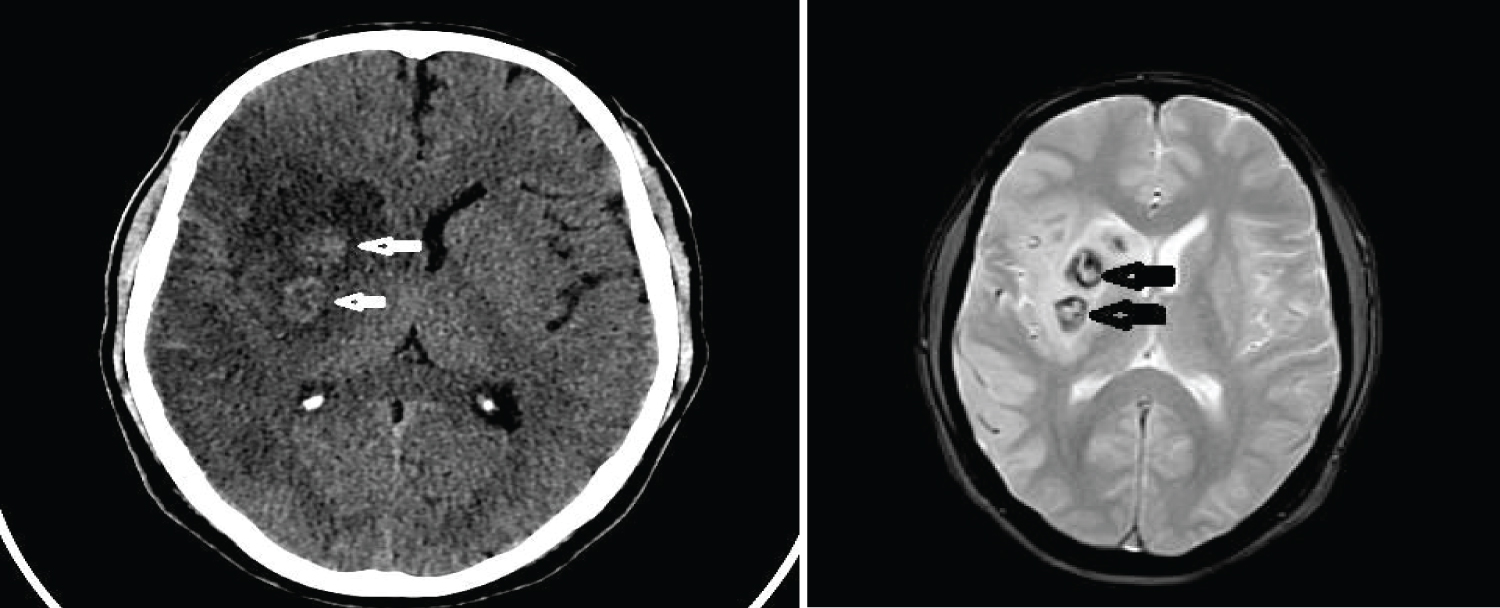 Figure 4: CT scan and MRI (FFE sequence) showing hemorrhagic transformation in right MCA Infarct.
View Figure Chart 4
Figure 4: CT scan and MRI (FFE sequence) showing hemorrhagic transformation in right MCA Infarct.
View Figure Chart 4
His echocardiogram showed a left ventricular apical aneurysm with mural thrombus. In view of the large infarct with hemorrhagic change and cerebral edema with midline shift it was decided to defer anticoagulation for the first 14 days post stroke.
A routine follow up MRI brain done on day 13 post stroke showed that blooming in the right basal ganglia had reduced and there was considerable reduction in edema and no midline shift, however he was noted to have thrombosis of the left transverse sinus, left sigmoid sinus and proximal part of the left internal jugular vein. He was asymptomatic with no headache and improving clinically at this time (Figure 5 and Figure 6).
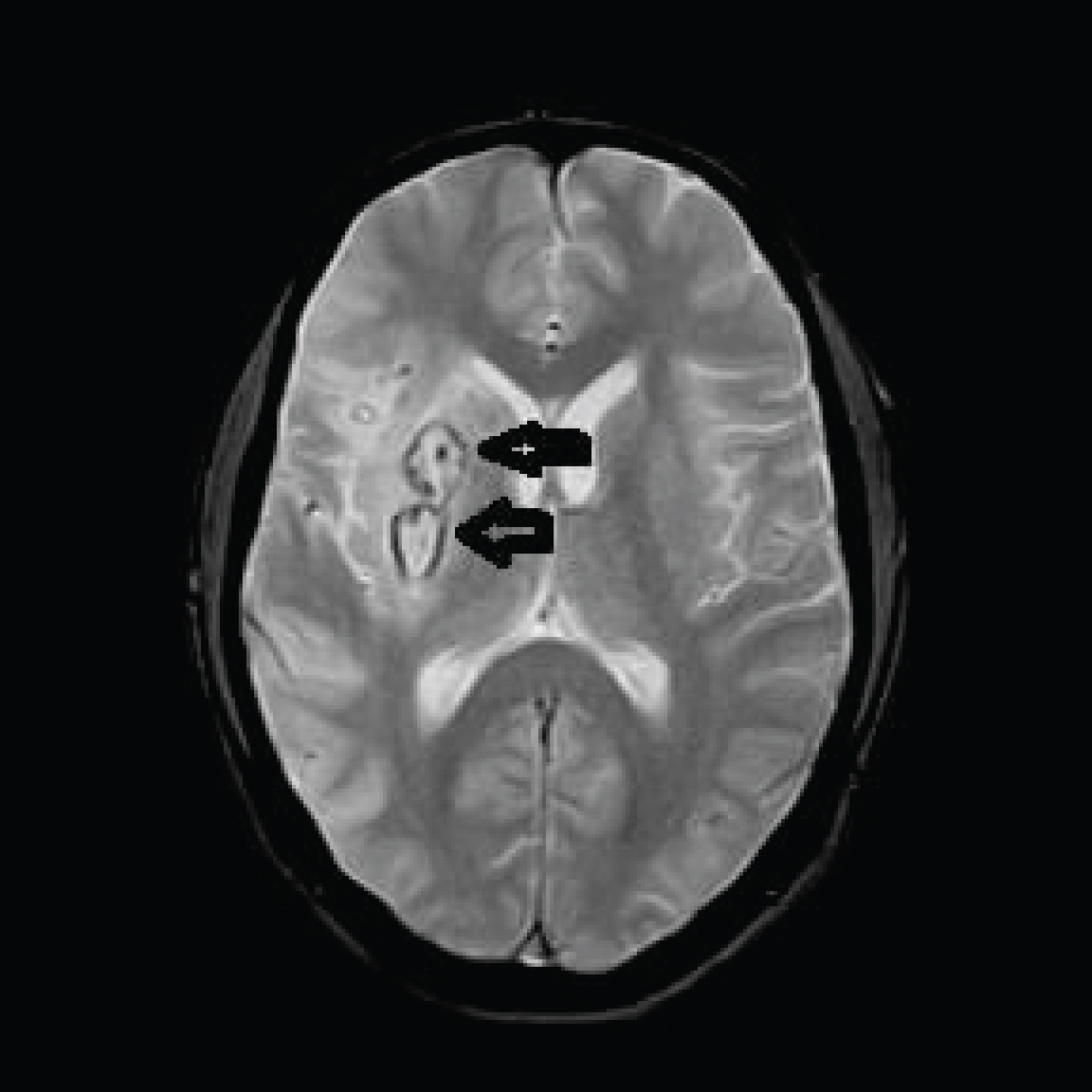 Figure 5: MR showing reduced Blooming and edema on post treatment follow up FFE sequence.
View Figure Chart 5
Figure 5: MR showing reduced Blooming and edema on post treatment follow up FFE sequence.
View Figure Chart 5
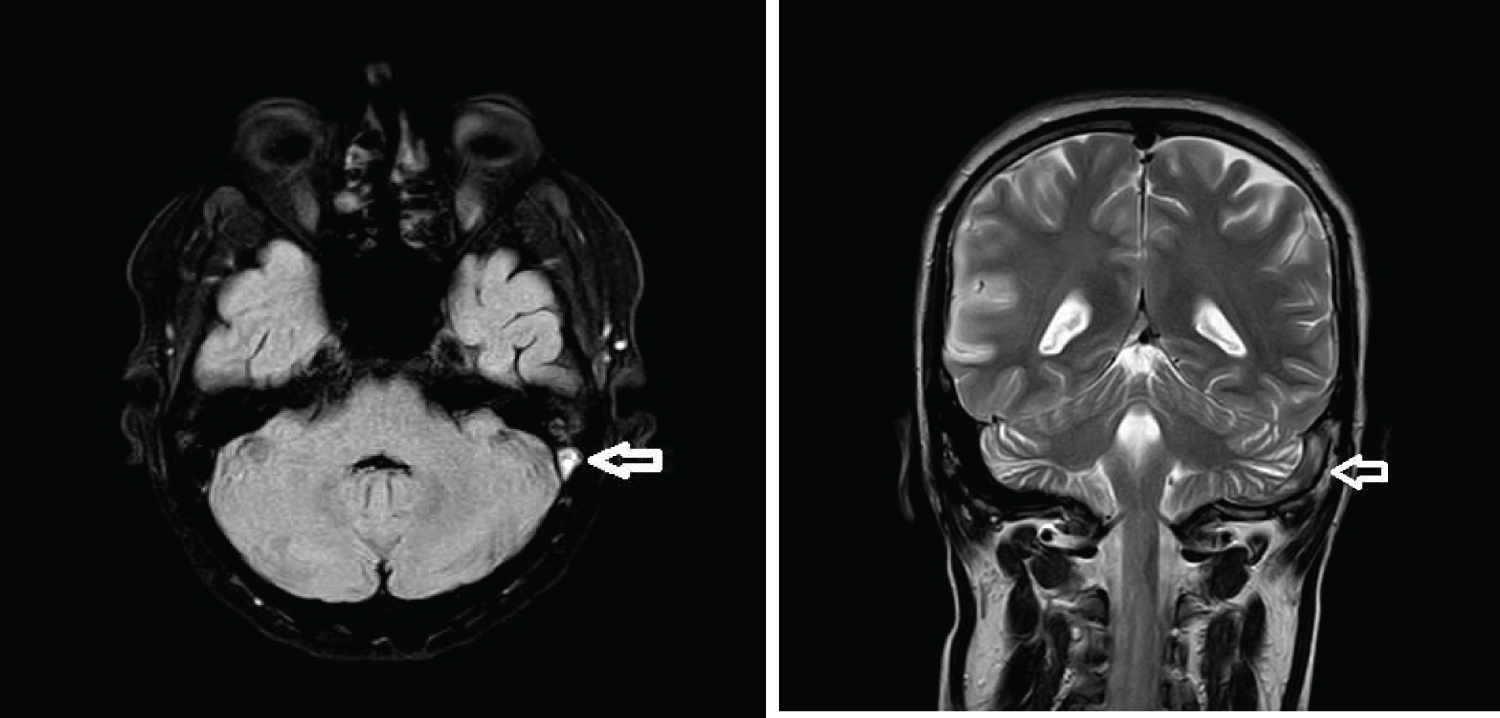 Figure 6: FLAIR axial and T2 coronal sequence show left transverse sinus thrombosis.
View Figure Chart 6
Figure 6: FLAIR axial and T2 coronal sequence show left transverse sinus thrombosis.
View Figure Chart 6
He was anticoagulated initially with fractionated heparin and later with warfarin with maintenance of an INR between 2 and 3. He made a good recovery and at discharge was walking independently with normal limb power.
Extensive thrombophilia and vasculitis work up had been sent prior to anticoagulation and this revealed significant Antithrombin 3 (AT3) deficiency (70% of normal activity).
Antithrombin III also called AT3, is a naturally occurring anticoagulant. Its function is to inhibit thrombin (factor IIa), factor Xa, and other serine proteases in the coagulation cascade such as factor IXa [1,2]. AT has a half-life of approximately 2.8 to 4.8 days.
AT deficiency can be genetic or acquired. Genetic deficiencies are due to AT gene mutations. Inheritance of AT deficiency is autosomal dominant with variable penetrance. Offspring of affected individuals have approximately a 50 percent chance of inheriting the condition. Many other mutations and other genetic alterations such as deletions have been identified in the gene encoding AT (SERPINC1) [3]. The resulting phenotypes are divided into type I and type II deficiencies. In Type I there is an actual deficiency of AT3, in type II although the amount of AT is normal, there is a dysfunctional alteration of AT3 [4].
Acquired deficiencies can result from impaired production of functional AT (eg, due to liver disease, asparaginase therapy); protein losses (eg, in nephrotic syndrome) or accelerated consumption, as occurs in acute thrombosis or disseminated intravascular coagulation (DIC).
Our patient had significant AT3 deficiency with 70% of normal activity only; any value below 80% of normal activity is considered AT3 deficiency. Testing had been done on day 11 post stroke before starting anticoagulation.
The AT3 deficiency was probably genetic as he had no etiology for acquired causes such as nephrotic syndrome [5], liver disease [6], hemodialysis or asparaginase therapy [7,8].
The significant feature in our patient was the arterial thrombosis with an intracardiac mural thrombus and subsequent embolic stroke, the link between arterial thrombosis and AT3 deficiency is at best weak or tenuous. AT3 deficiency is more often associated with venous thromboembolism which our subject also had in the form of dural sinus thrombosis in addition to the arterial thrombosis. It is to be noted he had also previously had a myocardial infarction at the age of 25 years.
The other unusual feature in our subject was the almost simultaneous occurrence of arterial and venous thromboembolism in the setting of AT3 deficiency.
To our knowledge this unusual occurrence has not been reported in the setting of AT3 deficiency. We advocate extensive testing for thrombophilic conditions including AT3 activity when there is simultaneous occurrence of arterial and venous thromboembolism irrespective of the existence of other conventional vascular risk factors.