

International Journal of Stem cell Research and Therapy
Stem Cell Approach to Generate Cancer Specific Immune Effectors Cells
Sylvie Shen1, Ning Xu1, Geoff Symonds2 and Alla Dolnikov1*
1Sydney Children's Hospital, Sydney, Australia
2Calimmune Inc., Australia
*Corresponding author: Alla Dolnikov, Sydney Children’s Hospital, High Street, Randwick, NSW 2031, Australia, Tel: 61-2-93821879, Fax: 61-2-93820372, E-mail: Alla.Dolnikov@sesiahs.health.nsw.gov.au
Int J Stem Cell Res Ther, IJSCRT-3-022, (Volume 3, Issue 1), Review Article; ISSN: 2469-570X
Received: October 26, 2015 | Accepted: February 01, 2016 | Published: February 04, 2016
Citation: Shen S, Xu N, Symonds G, Dolnikov A (2016) Stem Cell Approach to Generate Cancer Specific Immune Effectors Cells. Int J Stem Cell Res Ther 3:022. 10.23937/2469-570X/1410022
Copyright: © 2016 Shen S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The ability to target cancer cells using genetically enhanced immune effector cells armed with a chimeric antigen receptor (CAR) capable of recognising tumour associated antigen is a novel approach to the treatment of cancer. Remarkable success in early phase clinical trials has demonstrated a potent anti-cancer effect of T cells modified to express CAR (CART cells) targeting CD19+ B-cell malignancies. Therapy using CAR-modified T cells (CART cell therapy) is, however, associated with a variety of problems, including supraphysiologic CART cell proliferation often resulting in toxicity in the form of cytokine release syndrome (CRS). Additionally, an often limited persistence of infused CART cells in the blood of the treated patients results in tumour recurrence. By designing a therapy that provides more durable immune responses, CART cells potentially could be used as curative therapy. We and others have developed a novel approach to promote long-lasting tumour immunity using adoptive transfer of ex-vivo generated CAR-modified T-cell precursors which can provide a continuous source of mature anti-cancer T-cells. Introduction of CARs to hematopoietic stem cells (HSCs) is an alternative target for immunotherapy. HSCs have strong regenerative and self-renewal capacity and therefore can potentially provide patients with high levels of CART cells and other immune effector cells targeting cancer potentially throughout life.
Keywords
Haematopoietic stem cell, T cell, Chimeric antigen receptor
Chimeric Antigen Receptor for Cell Based Therapy
Targeting cancer cells with immune cells armed with artificial chimeric antigen receptor (CAR) is a novel approach with proven success in early phase clinical trials for patients with CD19+ B-cell malignancies [1-6]. CARs combine the antigen-binding properties of an antibody with a T cell signaling domain (Figure 1). CARs activate T cells upon interaction with cells expressing the respective target antigen on their surface. Patient's own or donor T cells can be genetically modified to express CAR, amplified ex-vivo to numbers suitable for adoptive cell therapy and administered to the patient. T cells expressing CAR (CART cells) traffic to tumour sites where they become activated upon engagement with the tumour antigen and develop cytolytic activity against tumour cells [1-6]. Different CAR designs are employed that include not only antibody-derived scFv recognition domain bound to the CD3ζ intracellular signalling domain as it is in the first generations of CARs, but also one or two co-stimulatory domains (second and third generations of CARs, respectively). Different gene transfer approaches including viral and non-viral vectors as well as RNA-based methods are used to modify T cells to express CARs [6]. Additionally, different methods have been developed to manufacture large numbers of CAR-modified T cells. These methods include anti-CD3/CD28 antibody-coated magnetic beads, artificial antigen presenting cells and specific cytokines that stimulate CART cell proliferation and maintain their memory phenotype and function.
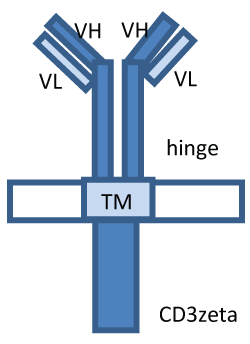
.
Figure 1: The structure of CAR. CARs consist of single-chain variable fragment (VL/VH) of tumour antigen-specific antibody fused to the transmembrane domain (TM) and CD3 zeta T cell activating signaling domain (CD3zeta).
View Figure 1
Ongoing clinical trials indicate that CART cells targeting CD19, an antigen expressed on leukaemia B cells, can be safely infused to patients and induce complete remission in a significant proportion of patients with B-cell leukaemia and lymphomas [1-7]. A number of challenges, however, still remain including minimising the severity of toxicities such as life-threatening cytokine release syndrome (CRS) often associated with the effective response to CART cell therapy. Additionally, it is appreciated that CD19 is not the ideal target since it is also expressed by normal B cells, and, therefore, the successful CART cell therapy targeting CD19 is always associated with chronic B-cell aplasia and hypogammaglobulinemia. B-cell aplasia, however, is treatable using immunoglobulin infusions that are tolerable and efficiently mitigate the infection complications [6]. Search for other potential targets may minimise on-target off-tumour side effect of CART cell therapy in future. Finally, relapse after CART-cell therapy occurs in significant proportion of patients with 30% relapsing by 6 months [6]. Sustained remission strongly correlates with CART cell expansion and persistence, with decreasing levels or loss of detectable CART cells in patients’ circulation frequently heralding imminent leukaemia relapse [3,6]. Therefore CART cell therapy is often used as a bridge to a stem cell transplant and not curative. By designing a product that provides durable immune responses CART cells could be used as curative therapy.
Strategies to Improve CART Cell Persistence
The mechanisms regulating in vivo expansion and sustainability of CART cells remain elusive. We have shown that CART cells targeting CD19 in chemoresistant B-cell acute lymphoblastic leukaemia (B-ALL) mediate potent in vitro cytolysis and show anti-tumour activity in immunodeficient xenograft models [8]. CAR T cells stimulated with CD19+ leukaemia cells derived from individual patients demonstrated variable degrees of expansion. CART cell expansion correlated with the expression of antigen targeted by CART cells [8]. It is relevant that down-regulation of CD19 was observed in the proportion of patients that relapsed after CART-cell therapy [1]. Selective pressure mediated by CART cell therapy appears to promote the development of CD19-negative leukaemia. Down-regulation of target antigen expression during clonal leukaemia evolution, therefore, may reduce CART-cell expansion and persistence in patients received CART-cell therapy.
Molecular profiling of individual B-ALL patient samples revealed significant variations in the levels of stimulatory and inhibitory immune ligand expression. Significant proportion of patients expressed low levels of stimulatory immune ligands CD80, CD86, CD40L, ICOSL and CD27L. Down-regulation of stimulatory immune ligands therefore appears to be common feature in B-ALL and may act to prevent the expansion of adoptively transferred CART cells. Addition of genes encoding immune stimulatory ligands such as CD80 or CD40L into CAR structure was shown to promote CART-cell expansion [9-11]. We have recently shown that pre-treatment of leukaemia cells with a DNA methyltransferase inhibitor azacytidine (AZA) acts to up-regulate CD80 expression and promotes CART-cell expansion [8]. Additionally, pre-treatment with AZA before CART-cell infusion leveraged the anti-tumour activity of CART cells in mice transplanted with leukaemia cells [8]. Providing additional co-stimulatory signalling, thus, appears to promote CART-cell expansion and improve their in vivo persistence.
Antigen-specific stimulation promotes CART cell proliferation and differentiation into short-lived effector cells expressing high levels of programmed cell death protein 1 (PD-1) and CTLA-4. The interaction between PD-1 and its ligands PD-L1 or PD-L2 inhibits proliferation and effector functions of T cells and induces apoptosis [12] while CTLA-4 engagement by its ligand CD80 or CD86 delivers negative signals that circumvent the CD28-mediated co-stimulatory signalling [13]. Upregulation of inhibitory ligands in tumour microenvironment may provide an immune escape for leukaemia cells by turning off the adoptively transferred CART cells. Improved anti-tumour responses were seen in a murine model that combined checkpoint inhibitor anti-PD1 with CART cells [14]. These data may justify the use of inhibitory receptors/ligands antagonists (checkpoint inhibitors) to promote CART cell expansion and improve their persistence.
CART cell exhaustion is further augmented by chronic CAR signalling mediated by high tumour burden [8]. In the presence of large numbers of target cells, repetitive antigen-specific stimulation induced extensive proliferation of CART cells that lead to the rapid differentiation into short-lived effector cells. Increasing effector to target ratio delayed CART cell differentiation and promoted their persistence. Consistent with this finding, cytoreductive treatments reducing tumour burden maximised CART cell immune responses [3].
Co-stimulatory signalling triggered by specific cytokines is critical for prolonged T cell activation, and inclusion of co-stimulatory domains such as CD28, 4-1BB, ICOS and OX40, increased CART cell expansion and improve in vivo persistence [15-18]. In CARs with the inbuilt co-stimulatory domains, antigen-specific stimulation activates co-stimulatory signalling independent on exogenous co-stimulatory ligands. Activation of co-stimulatory signalling provides additional pro-survival signal and delays CART cell extinction following encounter with tumour antigen. Using second generation of CARs with inbuilt CD28 co-stimulatory domain we have shown that CART cells efficiently kill leukaemia cells commonly expressing low levels of co-stimulatory ligand CD80 [8]. Composition of CARs has been optimised by including combination of two co-stimulatory molecules or combining co-stimulatory domains with the co-stimulatory ligands [15]. In addition, inclusion of genes encoding IL-12 and CD40L in CAR-construct prolonged CART cell activity [11,19]. Thus optimising CART cell composition by adding the genes encoding specific immune stimulatory ligands and/or cytokines can make CART cell function independent on immune suppressive tumour microenvironment and increase the efficacy of CART cell therapy particularly for patients with tumours expressing low levels of co-stimulatory ligands and cytokines.
CART Cells Generated from T cells with “Young” Phenotype
CART cells generated using patient’s mature T cells rapidly differentiate into short-lived effector memory (Tem) cells following antigen-specific stimulation [2,3,8]. Pre-clinical studies of adoptive T cell transfer have shown that although Tem have robust cytolytic function, T cells with younger phenotype – central memory T cells (Tcm) or naïve T cells (Tn) are critical for in vivo expansion and long-term persistence [20]. In addition, a rare subpopulation of T memory stem cells (Tmsc) has been shown to play an important role in supporting long-term efficacy of adoptive T cell therapy [21]. Ex vivo expansion of CART cells using γ-chain cytokines IL-7 and IL-15, acts to preserve Tcm and Tmsc subsets [22]. IL-15 promotes T cell survival by bcl-2 up-regulation, and confers resistance to the inhibitory effects of regulatory T cells [20]. We have recently shown that activation of the Wnt/β-catenin pathway using small molecule glycogen synthase kinase-3β (GSK3β) inhibition delayed Tn differentiation [23]. GSK3β inhibition down-regulates expression of genes activated during effector differentiation and preserves Tn gene expression. Accumulation of β-catenin, however, lead to a reduced CART cell expansion precluding obtaining sufficient T cells for adoptive therapies (unpublished data). Additionally, Akt inhibition was shown to enhance expansion of tumour specific T cells with memory characteristics [24]. Thus, pharmacologic inhibition of Akt represents additional approach to enhance the persistence of anti-tumour T cells.
A novel approach to improve mature CART cell persistence has been developed through the transduction of CAR into T cell precursors (pre-Ts) ex vivo generated from haematopoietic stem cells (HSCs). Pre-T cells generated from murine HSCs using conditions mimicking thymic microenvironment in co-culture with feeder OP9-DL1 cells activating Notch signalling were engineered to stably express first generation CAR targeting human CD19 (hCD19) and tested in allogeneic mouse model [25]. Adoptively transferred CAR-pre-T cells generated CD4+ and CD8+ CAR-expressing T cells and enhanced graft-versus-tumour activity in HSC recipients that had been challenged with hCD19+ tumour cells. These data indicate that CAR-engineered pre-T cells give rise to antigen-specific host-tolerant T cells that can display cytotoxic activity upon stimulation with their specific antigen, migrate to the site of antigen expression in vivo and persist for at least two months after transfer.
We have recently generated human pre-T cells using feeder cell free conditions with the addition of another Notch ligand-DL4 [26] and transduced them with the second generation CARs targeting hCD19. CAR-transduced human pre-Ts with CD34-CD7+ pro-T1 phenotype efficiently engrafted immunocompromised mice and generated mature T cells expressing CARs and demonstrating the enhanced anti-tumour activity in mouse leukaemia model. Collectively these data confirm that adoptive transfer of CAR-modified pre-T cells may represent an effective strategy for targeted immunotherapy for patients with malignant diseases.
Stem Cell based Approach to Generate CART Cells
Pre-Ts have limited self-renewal ability preventing long-lasting generation of CART cells. An alternative approach to promote long-lasting anti-tumour immunity is using haematopoietic stem cells (HSCs) modified with CARs (CAR-HSCs) [27]. Adoptive transfer of CAR-HSCs will generate continual supply of CART cells providing superior anti-cancer efficacy compared to infusion of CAR-modified pre-Ts or mature T cells (Figure 2). In addition, CAR expression will not be limited to T cells. CAR-HSCs will generate multiple immune effector cells such as neutrophils, monocytes and natural killer (NK) cells, which in addition to T cells, have demonstrated anti-leukaemia activity [28]. Importantly, generation of myeloid and NK cells does not depend on thymic function often impaired by chemotherapy in patients with cancer. CAR expression by multiple immune effector cells may confer combined and more robust anti-tumour immunity with continuously generated new effector cell types.
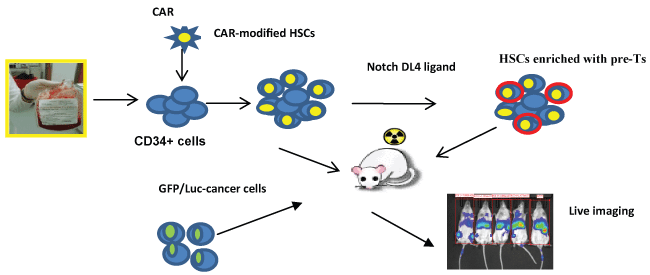
.
Figure 2: A schematic diagram of HSC approach to generate CAR-modified immune effector cells. Cord blood derived CD34+ HSCs cells are infected with lentivirus expressing CAR and infused to the sub-lethally irradiated NSG mice. Alternatively, mice can be infused with CAR-transduced CD34+ cells co-cultured with immobilized Notch ligand DL4 to promote T cell differentiation. Stem cell reconstituted mice are challenged with CD19+ leukaemia cells. Leukaemia progression is monitored by live imaging.
View Figure 2
Genetic modification of HSCs with CARs targeting CD19 antigen was recently reported [28]. Transgene expression was observed in the multilineage progeny of CAR-transduced cord blood derived CD34+ HSCs in vitro [28] and in immune-deficient NOD/SCID/IL2Rγ-null (NSG) mice transplanted with CAR-HSCs [28]. CAR gene transfer into HSCs did not impair hematopoietic differentiation and cell proliferation. NSG pups transplanted with CAR-HSCs presented CAR-bearing cells in haematopoietic tissues and gave rise to functional CAR-modified cells that specifically targeted hCD19 antigen. In vivo challenge of stem cell engrafted mice with CD19+ leukaemia cells demonstrated the inhibition of tumour development. These results provided the first evidence for the feasibility and efficacy of the modification of HSCs with CAR as a strategy for generating multiple lineages of immune effector cells for immunotherapy against B-cell malignancies to augment graft-versus-leukaemia (GVL) effect of allogeneic stem cell transplants.
We have also studied the reconstitution of multilineage haematopoiesis using human HSCs retrovirally transduced with CAR targeting hCD19. Additionally, we have transduced HSCs with CARs targeting GD2 antigen expressed by some solid tumours such as neuroblastoma and sarcomas. Transduction with each of these CARs achieved 60-70%, and CAR expression was seen in multilineage progeny of the transduced CD34+cells. CAR expression did not modulate stem cell proliferation and differentiation in vitro. Mice engrafted with hCD19-specific CAR-HSCs had decreased hCD19+ B-cell population confirming functionality of CAR targeting in vivo. In addition, delayed disease progression was observed in CAR-HSC-reconstituted mice challenged with hCD19+ Burkitt lymphoma Raji cells. Importantly, lymphoma burden inversely correlated with the proportion of T-cells confirming T-cell mediated anti-tumour effects. Further in vivo studies are needed to evaluate the effects of CAR expression of the cellular progeny such as non-specific cell activation, tissues homing and development of immunological memory. In summary, the data supporting the concept of genetic modification of HSCs with CARs is still very limited. With this initial progress towards CAR-modification of HSCs, further studies are needed to evaluate the most efficient approaches for CAR gene transfer to human HSCs. The optimised lentiviral vectors (LV) carrying a CAR should be evaluated for transgene expression and function in vivo. A major concern regarding the clinical use of LV vectors is the risk of insertional mutagenesis. Therefore the potential of LV-CAR-mediated mutagenesis must be assessed [29]. Importantly, to avoid potential insertional mutagenesis and make the approach clinically relevant, a self-inactivating LVs carrying a second generation CARs containing different co-stimulatory domains must be engineered to transduce human HSCs with CARs. The inducible CAR expression may be required to avoid potential cytotoxicity induced by combined immune effectors generated by CAR-HSCs [30]. The inclusion of the safety switch features to the vector to suppress CART cells if serious side effects emerge, or to make them self-destruct if they attack healthy tissue may improve the safety of this approach [31]. Further studies are needed to evaluate the biology and function of the different sub-populations of CAR-modified T cells produced by HSCs. It remains to be examined whether the diverse CAR-expressing immune effector cells will cause unique toxicities. In addition, the relative benefits of adding the activity of CAR-modified myeloid and NKs cells to the T-cells remain to be examined. Effects on cellular progeny need to be assessed such as non-specific cell activation, tissues homing and development of immunological memory.
Summary
Stem cell approach has been proposed to generate potent multi-lineage immune effector cells targeting cancer through genetic engineering of patient’s own or donor stem cells. This concept unites the combined cancer-killing power of multilineage immune cells with the long-term regenerative power of HSCs and represents a paradigm-shifting approach in the use of immunotherapy to treat cancer. Currently CAR therapy is used as a ‘bridge’ to transplant or an alternate therapy, providing short lived response rates in majority of patients. CAR-HSC approach will provide patients with a continual supply of genetically modified immune effectors cells precisely targeted toward their malignant cells. Not only can CAR-HSCs generate large numbers of anti-tumour T cells to continually control the disease, CAR-HSCs may prove more therapeutically relevant over mature CART cells since de-novo generated naïve CART cells will continuously replace the “used” CART cells. In addition, CAR-HSCs will provide patients with a combined supply of multilineage immune effector cells expressing CARs, thus enhancing the effect of CART cells. Use of CAR-HSCs to produce multiple immune effector cells with specific cytolytic activity could be employed in the context of bone marrow transplantation to augment the anti-tumour activity.
References
-
Maus MV, Grupp SA, Porter DL, June CH (2014) Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood 123: 2625-2635.
-
Kenderian SS, Ruella M, Gill S, Kalos M4 (2014) Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer Res 74: 6383-6389.
-
Gill S, June CH (2015) Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263: 68-89.
-
Xu XJ, Zhao HZ, Tang YM (2013) Efficacy and safety of adoptive immunotherapy using anti-CD19 chimeric antigen receptor transduced T-cells: a systematic review of phase I clinical trials. Leuk Lymphoma 54: 255-260.
-
Rambaldi A, Biagi E, Bonini C, Biondi A, Introna M1 (2015) Cell-based strategies to manage leukemia relapse: efficacy and feasibility of immunotherapy approaches. Leukemia 29: 1-10.
-
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, et al. (2014) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371: 1507-1517.
-
Maude SL, Teachey DT, Porter DL, Grupp SA3 (2015) CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125: 4017-4023.
-
Dolnikov A, Shen S, Klamer G, Joshi S, Xu N, et al. (2015) Anti-leukemic potency of CD19-specific T cells against chemo resistant paediatric acute lymphoblastic leukaemia. Exp Hematol 43: 1001-1014.
-
Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, et al. (2015) Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS One 10: e0130518.
-
Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, et al. (2015) Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Molecular Therapy 23: 769-78.
-
Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, et al. (2015) Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 28: 415-428.
-
Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, et al. (2014) Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20: 4262-4273.
-
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ , et al. (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1: 405-413.
-
John LB, Devaud C, Duong CP, Yong CS, Beavis PA, et al. (2013) Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 19: 5636-5646.
-
Zhang Q, Li H, Yang J, Li L, Zhang B, et al. (2013) Strategies to improve the clinical performance of chimeric antigen receptor-modified T cells for cancer. Curr Gene Ther 13: 65-70.
-
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, et al. (2009) Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 106: 3360-3365.
-
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, et al. (2011) CD28 co-stimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 121: 1822-1826.
-
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, et al. (2004) Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18: 676-684.
-
Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, et al. (2015) IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29: 415-422.
-
Barrett DM, Singh N, Liu X, Jiang S, June CH, et al. (2014) Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy16: 619-630.
-
Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, et al. (2013) Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest 123: 594-599.
-
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, et al. (2014) Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood 123: 3750-3759.
-
Shen S, Klamer G, Xu N, O'Brien TA, Dolnikov A (2013) GSK-3β inhibition preserves naive T cell phenotype in bone marrow reconstituted mice. Exp Hematol 41: 1016-1027.
-
Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, et al. (2015) Akt inhibition enhances expansion of potent tumor-specific lymphocytes with memory cell characteristics. Cancer Res 75: 296-305.
-
Zakrzewski JL, Suh D, Markley JC, Smith OM, King C, et al. (2008) Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat Biotechnol 26: 453-461.
-
Reimann C, Six E, Dal-Cortivo L, Schiavo A, Appourchaux K, et al. (2012) Human T-lymphoid progenitors generated in a feeder-cell-free Delta-like-4 culture system promote T-cell reconstitution in NOD/SCID/γc(-/-) mice. Stem Cells 30: 1771-1780
-
Gschweng E, De Oliveira S, Kohn DB (2014) Hematopoietic stem cells for cancer immunotherapy. Immunol Rev 257: 237-249.
-
De Oliveira SN, Ryan C, Giannoni F, Hardee CL, Tremcinska I, et al. (2013) Modification of hematopoietic stem/progenitor cells with CD19-specific chimeric antigen receptors as a novel approach for cancer immunotherapy. Hum Gene Ther 24: 824-839.
-
Carbonaro DA, Zhang L, Jin X, Montiel-Equihua C, Geiger S, et al. (2014) Preclinical demonstration of lentiviral vector-mediated correction of immunological and metabolic abnormalities in models of adenosine deaminase deficiency Mol Ther 22: 607-22.
-
Hoseini SS, Hapke M, Herbst J, Wedekind D, Baumann R, et al. (2015) Inducible T-cell receptor expression in precursor T cells for leukemia control. Leukemia 29: 1530-1542.
-
Gargett T, Brown MP2 (2014) The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol 5: 235.
