

International Journal of Stem cell Research and Therapy
Mesenchymal Stem Cell Modulation of TH17 Cells
Justin D Glenn1 and Katharine A Whartenby1,2*
1Department of Neurology, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States of America
2Department of Oncology, Johns Hopkins University School of Medicine, Baltimore, Maryland, United States of America
*Corresponding author: Katharine A Whartenby, PhD, Departments of Neurology and Oncology, Johns Hopkins School of Medicine, 601 N. Wolfe Street, Baltimore, Maryland, MD 21287, United States of America, Tel: +1-410-5023290, Fax: +1-443-2874062, E-mail: whartenby@jhmi.edu
Int J Stem Cell Res Ther, IJSCRT-3-035, (Volume 3, Issue 1), Short Review; ISSN: 2469-570X
Received: April 28, 2016 | Accepted: June 22, 2016 | Published: June 26, 2016
Citation: Glenn JD, Whartenby KA (2016) Mesenchymal Stem Cell Modulation of TH17 Cells. Int J Stem Cell Res Ther 3:035. 10.23937/2469-570X/1410035
Copyright: © 2016 Glenn JD, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Mesenchymal stem cells (MSCs) are multi-faceted cells capable of tissue regeneration, wound healing, and immunosuppression. Their immunosuppressive actions extend to most innate and adaptive immune cells, including TH17 cells, which have recently been discovered to be important pathogenic cells in a variety of disease settings, including many autoimmune diseases. As various long-standing treatments and therapies in autoimmune disease may face limitations and result in dangerous side effects, the field has searched for safer immunosuppressive therapies, and MSC-based therapy may hold great potential in controlling chronic TH17-based inflammatory conditions.
Keywords
Mesenchymal stem cells (MSCs), Inflammation, TH17
Short Review
Mesenchymal stem cells
Mesenchymal stem cells compose a heterogeneous stem cell population of the mesenchyme that exhibit multiple functions that make them appealing to different fields of biomedical and clinical research and application. Despite heterogeneity, they have some distinctive, shared features including phenotypic characteristics such as the lack of key hematopoietic molecular markers but expression of cluster of differentiation (CD) 90, CD105, CD44, CD73, CD9 and low levels of CD80 [1-3]. They were initially identified as self-renewing cells with fibroblastic appearance found in various organs and tissues, especially in the bone marrow, where they are thought to function physiologically as regulators of hematopoietic stem cell proliferation and differentiation [4-6]. These cells produce numerous growth factors and promote wound healing and angiogenesis [1,7]. In the presence of different molecular milieus, MSCs may differentiate into adipocytes, chondrocytes and osteoblasts.
In the 1970s, Friedenstein and colleagues described the ability of splenic fibroblasts to support hematopoietic microenvironments in transplantation experiments [8], suggesting some overlap between fibroblasts and MSCs. Fibroblast cells (which express vimentin, collagen prolyl 4-hydroxylase, and fibronectin) derived from human adult bronchi strongly express the MSC markers CD73, CD105 and CD166, lack expression of CD31, CD34, and CD45, and remarkably were able to undergo adipogenesis, chondrogenesis and osteogenesis under inducible conditions in culture [9]. Furthermore dermal skin-derived fibroblasts display similar MSC marker surface antigen phenotypes, can differentiate into bone, cartilage, and fat, and have high proliferation potential [10]. Other reports have since shown many similarities and very few differences between the two cell populations [11-13]. Covas and colleagues even revealed that fibroblasts and MSCs share a global gene expression pattern [14]. Thus there is still controversy concerning whether MSCs and fibroblasts are the same cell population, are derived from a common or similar progenitor, or represent a diverse continuum of different cells along a differentiation pathway that retain fundamental phenotypic and functional characteristics. For the purpose of this review, we will focus on primary literature based on investigation with cells with MSC nomenclature.
MSCs were initially evaluated for their potential to stimulate regenerative cellular and molecular pathways in degenerative disorders of mesenchymal origin [15]. Early studies demonstrated that the presence of MSCs greatly improved bone marrow transplantation and engraftment, and revealed another action of MSCs which contributed to decreased graft rejection-immunosuppression [16,17]. The basis of MSC immunosuppression stems from their physiological role of maintaining homeostasis of the hematopoietic stem cell (HSC)niche in the bone marrow, as inflammatory events may cause unsuspected HSC activation, differentiation, and depletion of HSC reservoirs [1]. In the HSC niche, MSC-derived fibroblasts have been shown to eliminate survival factors important for immune cells and to re-calibrate chemokine gradients, which may limit the presence of T cells, when studied in fibroblast dysfunction in the autoimmune disease rheumatoid arthritis [18]. MSCs also migrate from their normal anatomical sites to areas of inflammation to suppress activated T cell proliferation and cytokine production [19,20]. In line with the ultimate MSC physiological goal of maintaining homeostasis, suppression of inflammation permits the proliferation of tissue-resident stromal and stem cells of parenchymal tissue, production of extracellular matrix molecules for extracellular tissue reconstruction, and wound healing.
TH17 cells
CD4+ T cells are principle adaptive immune cells that function to coordinate and optimize multi-cellular immune responses and are activated as a function of events perceived as dangerous by the body. Antigen-presenting cells (APCs) such as dendritic cells (DCs) express many classes of pattern recognition receptors that bind endogenous, usually intracellular molecules released from cells during stress and necrosis and molecules derived from foreign, non-self-agents such as viruses, bacteria, and fungi [21]. In response to “dangerous” molecule-receptor binding and signaling, DCs up-regulate major histocompatibility complex (MHC) molecules for increased peptide binding and presentation, enhance T cell co-stimulatory molecule expression, and produce cytokines that, depending on composition and concentration, determine the effector function of the cognate CD4+ T cell. However, they may also mount inappropriate responses against self-tissue, as is the case in autoimmune pathogenesis. Historically, CD4+ T cells were considered to adopt one of two effector phenotypes that were thought to tailor the ensuing immune response to eliminate pathogens based on location in relation the very cells [22]. TH1 cells are generated in the presence of the pro-proliferative cytokine interleukin (IL)-2 and the inflammatory cytokine IL-12 and promote immunity to intracellular pathogens such as bacteria or viruses, while TH2 cells develop in the presence of IL-4 and promote immunity to extracellular pathogens including protozoa and fungi. The molecules that favor the effector phenotype of each antagonize the adoption of the other, and this axis was believed to be exclusive until the discovery of CD4+ T cells producing the highly inflammatory cytokine IL-17A, now known as TH17 cells [23]. TH17 cells are distinct from the aforementioned T cell populations at the transcriptional level by the expression of the transcription factor retinoic acid receptor (RAR)-related orphan receptor gamma t (RORγt); the TH1 effector program is directed by the T-box transcription factor T-bet and that of TH2 by GATA-binding protein 3 (GATA-3). TH17 cells develop in the presence of IL-6 and transforming growth factor-beta (TGF-β) and, in addition to IL-17A, produce the cytokines IL-17F, IL-21, and IL-22. The TH17 program is expanded and stabilized by the signaling of IL-23. Initial deductive studies of cytokines that promoted TH17 development were done with murine CD4+ T cells and later studies have shown that IL-6 in combination with IL-1 and IL-23, but not with TGF-β, can promote human TH17 development [24]. The in vivo role of TGF-β in promoting human TH17 remains controversial [25,26]. TH17 cells are important in promoting immunity to highly pathogenic bacteria and fungi and are found in high frequencies in the gut to prevent bacterial translocation past the intestinal wall [27].
Since the discovery of TH1, TH2, and TH17, a variety of other effector CD4+ T cell types have been identified based on transcription factor expression, cytokines they produce, and the molecular milieu that favor generation of each lineage. IL-4 and TGF-β induce IL-9-producing TH9 cells in vitro, are marked by PU.1 expression, and are thought to be important in promoting allergic inflammation, autoimmune disease, and immunity to tumors [28]. TH22 cells are distinguished by IL-22 production (lack IFNγ and IL-17A expression), aryl hydrocarbon receptor expression, and develop in the presence of IL-6 and TNFα; they home to the skin and may protect against microbial pathogens as well promote tissue repair and remodeling [29]. These cells may also be involved in inflammatory conditions of the skin. Follicular helper T cells (Tfh) express the transcription factor Bcl-6 and home to B cell follicles and germinal centersto provide B cell help [30]. Tfh are thought to develop in the presence of IL-6, IL-21, and IL-12. Interestingly, these cells produce IL-4, IL-17, and IL-10 [31,32]. Regulatory CD4+ T cells (Treg) are generated physiologically and are induced in the periphery to resolve inflammation mainly via suppression of immune cell activation and inflammatory cytokine production. The most recognized lineage is defined by master Forkhead box transcription factor Foxp3 expression and production of anti-inflammatory TGF-β expression, which also promotes its development. However, other Foxp3-negative CD4+ T cell types have been identified, including Tr1 and Tr35 cells. Tr1 cells suppress immune responses by high production of IL-10 but to date have no unique master transcription factor governing lineage development [33]. Little is known about the development of Tr35 cells though this population powerfully suppresses immune responses via IL-35 production [34]. Although these cell groups are usually presented as stable, static effector populations, considerable overlap in master transcription factor expression and cytokine production has been demonstrated. Effector populations also exhibit plasticity, in which one effector population may be converted into another; this phenomenon has mostly been attributed to epigenetic modifications that modify gene transcription when T cells are transferred into a different phenotype-skewing milieu [30].
TH17 are important mediators of inflammation in many inflammatory diseases, especially chronic autoimmune diseases such as rheumatoid arthritis, pancreatitis, autoimmune diabetes, and multiple sclerosis (MS). Pre-clinical models of many of these autoimmune diseases show that these cells are important in the pathogenesis. Some of their functions that have been identified include the mobilization of cells and induction of cytokine and chemokine production, in large part from cells of the innate immune system, including neutrophils and monocytes. An additional critical function for central nervous system (CNS) pathology is their association with permeabilization of the blood-brain barrier.[26]. For example, people with MS have been shown to have a higher frequency of TH17 cells in cerebrospinal fluid than matched counterparts without MS [35,36], indicating a flow into and/or expansion of these cells within the CNS. As a further indication of potential pathology the frequency of these TH17 cells is also higher during relapses than during remissions. People with other autoimmune diseases such as systemic lupus erythematosus (SLE) have also been shown to have similar patterns, with have higher serum levels of IL-17 and TH17 cells [37]. Circulating β-cell specific TH17 cells are also greater in frequency in type-1 diabetes than in healthy controls as well [38]. TH17 cells therefore are attractive targets for therapy in chronic inflammatory diseases.
MSCs and TH17 modulation
Since the linkage of TH17 cell appearance in many autoimmune diseases and proof-of-concept pre-clinical studies demonstrating their contributions to pathogenesis, biomedical research has been keen on targeting these cells for therapeutic benefit [1]. Considering the known strong immunosuppressive actions exerted by MSCs on T cells and their safety in early clinical disease trials, MSCs were investigated for their potential modulation of TH17 development and improvement of inflammatory disease.
Numerous investigations have now demonstrated that MSCs potently suppress TH17 via myriad mechanisms (Figure 1). At an early stage of the immune response, MSCs can inhibit differentiation and function of monocyte-derived DCs, thereby inhibiting any further T cell activation. CD14+ human monocytes differentiated into DCs with granulocyte-macrophage colony stimulation factor (GM-CSF) and IL-4 in the presence of MSCs maintain CD14 expression, exhibit decreased endocytic capacity, and do not upregulate the T cell co-stimulatory markers CD80 and CD86 [39]. MSCs were also shown to decrease antigen presentation and co-stimulatory markers on fully differentiated DCs. MSCs may also impart a regulatory phenotype on DCs. Splenic stromal cells can induce nitric oxide (NO) production from DCs in a manner dependent on fibronectin [40,41]. These NO-producing DCs then suppressed T cell proliferation. In a recent study MSCs and their extracellular vesicles (EVs) were shown to induce an immature regulatory phenotype on DCs marked by IL-6 and IL-10 expression in co-culture [42]. MSC and MSC-EV-conditioned glutamate decarboxylase 65 (GAD65)-pulsed DCs decreased antigen specific TH17 numbers and IL-17 secretion and promoted Treg development in the context of type 1 diabetes [42].
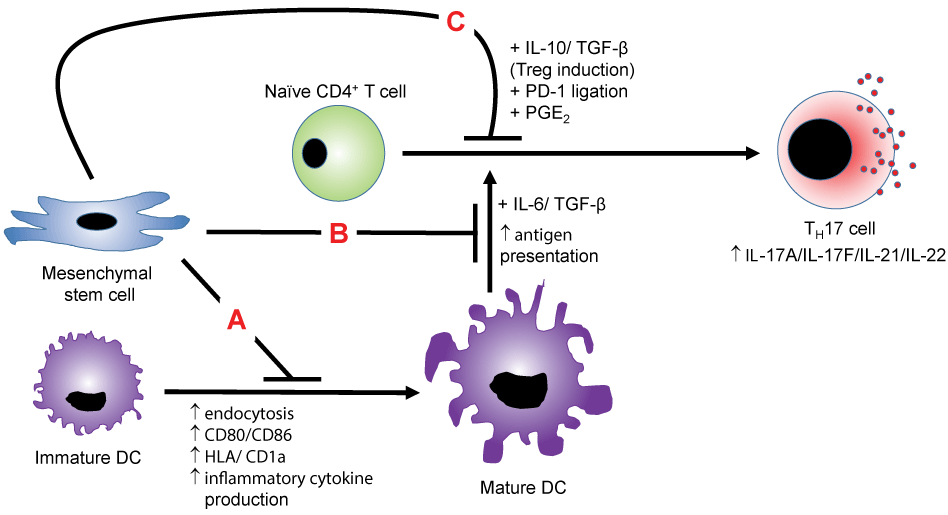
.
Figure 1: MSC suppression of TH17 cells-MSCs suppress the polarization of naïve CD4+ T cells towards TH17 cells via multiple mechanisms. In mechanism A, MSCs suppress the maturation of the antigen-presenting DC and also inhibit the presentation process to naïve CD4+ T cells in mechanism B, thereby indirectly preventing TH17 development. In mechanism C, MSC production of IL-10 and TGF-β redirects the polarization towards the immunosuppressive Treg cell type. MSCs may also inhibit T cell function via PD-1 ligation and PGE2 production.
View Figure 1
In several studies, MSCs have been shown to divert the development of naïve human CD4+ T cells away from the IL-17A-producing program into the development of a regulatory CD4+ T cell phenotype characterized by the expression Foxp3 and IL-10 production. Importantly for disease applications, MSCs could also induce Treg conversion from fully differentiated TH17 [43-45]. Reduction of full T cell activation was associated with signaling through the inhibitory cell surface receptor Programmed Death-1 (PD-1) on T cells and reduction of IL-17A expression was associated with increased expression of the immunosuppressive cytokine IL-10 [46]. MSCs co-cultured with these cells showed heightened transcription of TGF-β and prostaglandin E-2 (PGE2), with reversion of MSC suppression of the TH17 program made possible by a selective EP4 antagonist [47]. However, it has also been shown that MSCs are less effective at preventing IL-17A production with more fully differentiated TH17 cells, which contradicts the previous observation. Such differences could be due to a number of factors, including the conditions under which the TH17 cells were generated that may influence the stability of the phenotype. For example, TH17 cells exposed to IL-23 during the expansion phase have been shown to be more resistant to Tregthan those expanded without this cytokine [25,43]. While most studies have focused on the CD4+ lineage, CD8+ T cells may also produce IL-17A and have thus been termed Tc17 cells. Tc17 cells likely play a significant role in MS pathogenesis as they are present in MS lesions. Interestingly, they have been shown to also be susceptible to MSC suppression, via early enhanced production of IL-2 [48].
While there are numerous investigations supporting a suppressive role of MSCs in TH17 development, others have reported conflicting results. For example, MSC-derived cells, such as fibroblasts, may actually promote TH17 generation. IL-1β and TNFα derived from LPS-activated DCs stimulate human fibroblasts to produce PGE2, which enhances IL-23 production from DCs and increases TH17 cell development as well as IFNγ production [49,50]. Discrepancies in the effects of MSC action involving PGE2 may relate to the dose-dependent effect of PGE2 on the indoleamine-2,3-dioxygenase IDO pathway in DCs. PGE2 stimulates IDO activity in DCs and translates to their stimulation of allogeneic T cell proliferation, but inhibits DC ability to stimulate these T cells at high concentrations. This impacts TH1, TH2, and TH17 development [51].
In moving towards use of MSCs in therapeutic settings for TH17-mediated diseases, some pre-clinical murine experimental models of disease have shown potential benefit of MSC therapy for TH17-mediated diseases. For example, MSCs have been shown to ameliorate experimental autoimmune encephalomyelitis (EAE), a murine model of MS, via TH17 suppression and concomitant Treg induction [43,52]. MSCs were also shown to reverse multi-organ dysfunction in a mouse model of (SLE) via modulation of the TH17/Treg axis in favor of Treg cells [53]. These results indicate the potential for therapy in the future. However, several factors during the TH17 developmental stage must be considered. As stated, MSCs more effectively suppress IL-17A expression if the CD4+ T cells are exposed to MSCs during the T cell polarization phase. The MSCs both indirectly suppress T cells via APC function and directly by inhibiting T cell activation. In other words, when TH17 cells have already been generated, their suppression may become more difficult. These observations have been made mostly through the use of in vitro systems and do not account for anatomical barriers, multi-cellular complexities of tissues that may affect MSCs, and potential MSC differentiation. For example, MSCs can ameliorate the severity of MOG35-55 EAE when administered during the T cell priming phase and peak of disease but fail to alleviate the disease during stabilization [54]. MSCs in this study homed to lymphoid organs and the subarachnoid spaces. It may be the case that once encephalitogenic T cells have invaded the CNS parenchyma, they are out of reach of administered MSCs. On the contrary, MSCs restored the Treg/TH17 balance in a murine model of SLE when given in early and matured states of disease and reversed multi-organ dysfunction. In this model, MSCs were observed in the spleen and bone marrow [53]. These scenarios show that the effectiveness of MSC therapy may depend on stage of disease and anatomical compartmentalization of TH17 cells. Furthermore, differences in cytokine combinations and concentrations may affect MSC suppression in any given tissue. Further, there remains a dearth of knowledge on the fate of MSCs in various disease contexts. MSCs transdifferentiate into neural stem cells, mature neurons or glia in injured spinal cord tissue and may be able to form mature lung, gut epithelial, and muscle cells [55,56]. Transdifferentiation capacity is of utmost importance considering the TH17 promoting effect of MSC-derived fibroblasts and stromal cells and the potential loss of immunosuppression due to parenchymal cell formation.
MSCs have a large number of appealing features that could be exploited for therapeutic uses. Numerous studies show their potential for effective use, and molecular investigations into the mechanisms underlying MSC suppression of TH17 have revealed possible routes by which they might function. Taken together, the results of these studies provide great hope for the movement of MSCs into clinical trials for treating chronic inflammatory conditions. The pluripotent nature of the cells is a double-edged sword, however, and with additional conflicting studies demonstrating possible adverse outcomes, caution should be taken regarding the individual usage in both disease-specific and timing specific scenarios. Ongoing investigations into these potent cells are likely to reveal further mechanistic information that will help in refining their future use to reach their full therapeutic potential.
References
-
Glenn JD, Whartenby KA (2014) Mesenchymal stem cells: Emerging mechanisms of immunomodulation and therapy. World J Stem Cells 6: 526-539.
-
Boxall SA, Jones E (2012) Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int 2012: 975871.
-
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, et al. (2006) Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8: 315-317.
-
Berman L, Stulberg Cs, Ruddle Fh (1955) Long-term tissue culture of human bone marrow. I. Report of isolation of a strain of cells resembling epithelial cells from bone marrow of a patient with carcinoma of the lung. Blood 10: 896-911.
-
Mcculloch Ea, Parker Rc (1957) Continuous cultivation of cells of hemic origin. Proc Can Cancer Conf 2: 152-167.
-
Friedenstein AJ, Chailakhjan RK, Lalykina KS (1970) The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet 3: 393-403.
-
Seppanen E, Roy E, Ellis R, Bou-Gharios G, Fisk NM, et al. (2013) Distant mesenchymal progenitors contribute to skin wound healing and produce collagen: evidence from a murine fetal microchimerism model. PLoS One 8: e62662.
-
Friedenstein AJ, Chailakhyan RK, Latsinik NV, Panasyuk AF, Keiliss-Borok IV (1974) Stromal cells responsible for transferring the microenvironment of the hemopoietic tissues. Cloning in vitro and retransplantation in vivo. Transplantation 17: 331-340.
-
Sabatini F, Petecchia L, Tavian M, Jodon de Villeroché V, Rossi GA, et al. (2005) Human bronchial fibroblasts exhibit a mesenchymal stem cell phenotype and multilineage differentiating potentialities. Lab Invest 85: 962-971.
-
Lorenz K, Sicker M, Schmelzer E, Rupf T, Salvetter J, et al. (2008) Multilineage differentiation potential of human dermal skin-derived fibroblasts. Exp Dermatol 17: 925-932.
-
Lysy PA, Smets F, Sibille C, Najimi M, Sokal EM (2007) Human skin fibroblasts: From mesodermal to hepatocyte-like differentiation. Hepatology 46: 1574-1585.
-
Blasi A, Martino C, Balducci L, Saldarelli M, Soleti A, et al. (2011) Dermal fibroblasts display similar phenotypic and differentiation capacity to fat-derived mesenchymal stem cells, but differ in anti-inflammatory and angiogenic potential. Vasc Cell 3: 5.
-
Cappellesso-Fleury S, Puissant-Lubrano B, Apoil PA, Titeux M, Winterton P, et al. (2010) Human fibroblasts share immunosuppressive properties with bone marrow mesenchymal stem cells. J Clin Immunol 30: 607-619.
-
Covas DT, Panepucci RA, Fontes AM, Silva WA Jr, Orellana MD, et al. (2008) Multipotent mesenchymal stromal cells obtained from diverse human tissues share functional properties and gene-expression profile with CD146+ perivascular cells and fibroblasts. Exp Hematol 36: 642-654.
-
Wei X, Yang X, Han ZP, Qu FF, Shao L, et al. (2013) Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol Sin 34: 747-754.
-
Müller I, Kordowich S, Holzwarth C, Isensee G, Lang P, et al. (2008) Application of multipotent mesenchymal stromal cells in pediatric patients following allogeneic stem cell transplantation. Blood Cells Mol Dis 40: 25-32.
-
Prasad VK, Lucas KG, Kleiner GI, Talano JA, Jacobsohn D, et al. (2011) Efficacy and safety of ex vivo cultured adult human mesenchymal stem cells (Prochymal) in pediatric patients with severe refractory acute graft-versus-host disease in a compassionate use study. Biol Blood Marrow Transplant 17: 534-541.
-
Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, et al. (2001) Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol 22: 199-204.
-
Kolf CM, Cho E, Tuan RS (2007) Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res Ther 9: 204.
-
Shi Y, Hu G, Su J, Li W, Chen Q, et al. (2010) Mesenchymal stem cells: a new strategy for immunosuppression and tissue repair. Cell Res 20: 510-518.
-
Brown BD, Lillicrap D (2002) Dangerous liaisons: the role of "danger" signals in the immune response to gene therapy. Blood 100: 1133-1140.
-
Kidd P (2003) Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev 8: 223-246.
-
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27: 485-517.
-
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, et al. (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467: 967-971.
-
Romagnani S, Maggi E, Liotta F, Cosmi L, Annunziato F (2009) Properties and origin of human Th17 cells. Mol Immunol 47: 3-7.
-
Patel DD, Kuchroo VK (2015) Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 43: 1040-1051.
-
Huber S, Gagliani N, Flavell RA (2012) Life, death, and miracles: Th17 cells in the intestine. Eur J Immunol 42: 2238-2245.
-
Kaplan MH (2013) Th9 cells: differentiation and disease. Immunol Rev 252: 104-115.
-
Mirshafiey A, Simhag A, El Rouby NM, Azizi G (2015) T-helper 22 cells as a new player in chronic inflammatory skin disorders. Int J Dermatol 54: 880-888.
-
Bonelli M, Shih HY, Hirahara K, Singelton K, Laurence A, et al. (2014) Helper T cell plasticity: impact of extrinsic and intrinsic signals on transcriptomes and epigenomes. Curr Top Microbiol Immunol 381: 279-326.
-
Crotty S (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29: 621-663.
-
Reinhardt RL, Liang HE, Locksley RM (2009) Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10: 385-393.
-
Zeng H, Zhang R, Jin B, Chen L (2015) Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol 12: 566-571.
-
Spoerl S, Li XC (2011) Regulatory T cells and the quest for transplant tolerance. Discov Med 11: 25-34.
-
Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E (2009) Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 132: 3329-3341.
-
Li Y, Wang H, Long Y, Lu Z, Hu X (2011) Increased memory Th17 cells in patients with neuromyelitis optica and multiple sclerosis. J Neuroimmunol 234: 155-160.
-
Wong CK, Lit LC, Tam LS, Li EK, Wong PT, et al. (2008) Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol 127: 385-393.
-
Arif S, Moore F, Marks K, Bouckenooghe T, Dayan CM, et al. (2011) Peripheral and islet interleukin-17 pathway activation characterizes human autoimmune diabetes and promotes cytokine-mediated beta-cell death. Diabetes 60: 2112-2119.
-
Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, et al. (2005) Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood 105: 4120-4126.
-
Zhang M, Tang H, Guo Z, An H, Zhu X, et al. (2004) Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol 5: 1124-1133.
-
Flavell SJ, Hou TZ, Lax S, Filer AD, Salmon M, et al. (2008) Fibroblasts as novel therapeutic targets in chronic inflammation. Br J Pharmacol 153 Suppl 1: S241-246.
-
Favaro E, Carpanetto A, Caorsi C, Giovarelli M, Angelini C, et al. (2016) Human mesenchymal stem cells and derived extracellular vesicles induce regulatory dendritic cells in type 1 diabetic patients. Diabetologia 59: 325-333.
-
Luz-Crawford P, Kurte M, Bravo-Alegría J, Contreras R, Nova-Lamperti E, et al. (2013) Mesenchymal stem cells generate a CD4+CD25+Foxp3+ regulatory T cell population during the differentiation process of Th1 and Th17 cells. Stem Cell Res Ther 4: 65.
-
Obermajer N, Popp FC, Soeder Y, Haarer J, Geissler EK, et al. (2014) Conversion of Th17 into IL-17A(neg) regulatory T cells: a novel mechanism in prolonged allograft survival promoted by mesenchymal stem cell-supported minimized immunosuppressive therapy. J Immunol 193: 4988-4999.
-
Ghannam S, Pène J, Moquet-Torcy G, Jorgensen C, Yssel H (2010) Mesenchymal stem cells inhibit human Th17 cell differentiation and function and induce a T regulatory cell phenotype. J Immunol 185: 302-312.
-
Luz-Crawford P, Noël D, Fernandez X, Khoury M, Figueroa F, et al. (2012) Mesenchymal stem cells repress Th17 molecular program through the PD-1 pathway. PLoS One 7: e45272.
-
Duffy MM, Pindjakova J, Hanley SA, McCarthy C, Weidhofer GA, et al. (2011) Mesenchymal stem cell inhibition of T-helper 17 cell- differentiation is triggered by cell-cell contact and mediated by prostaglandin E2 via the EP4 receptor. Eur J Immunol 41: 2840-2851.
-
Glenn JD (2014) Mesenchymal stem cells differentially modulate effector CD8+ T cell subsets and exacerbate experimental autoimmune encephalomyelitis. Stem Cells 32: 2744-2755.
-
Schirmer C, Klein C, von Bergen M, Simon JC, Saalbach A (2010) Human fibroblasts support the expansion of IL-17-producing T cells via up-regulation of IL-23 production by dendritic cells. Blood 116: 1715-1725.
-
Malecka A, Wang Q, Shah S, Sutavani RV, Spendlove I, et al. (2016) Stromal fibroblasts support dendritic cells to maintain IL-23/Th17 responses after exposure to ionizing radiation. J Leukoc Biol.
-
Lanzinger M, Jürgens B, Hainz U, Dillinger B, Raberger J, et al. (2012) Ambivalent effects of dendritic cells displaying prostaglandin E2-induced indoleamine 2,3-dioxygenase. Eur J Immunol 42: 1117-1128.
-
Rafei M (2009) Mesenchymal stromal cells ameliorate experimental autoimmune encephalomyelitis by inhibiting CD4 Th17 T cells in a CC chemokine ligand 2-dependent manner. J Immunol 182: 5994-6002.
-
Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, et al. (2009) Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells 27: 1421-1432.
-
Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, et al. (2005) Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood 106: 1755-1761.
-
Satake K, Lou J, Lenke LG (2004) Migration of mesenchymal stem cells through cerebrospinal fluid into injured spinal cord tissue. Spine (Phila Pa 1976) 29: 1971-1979.
-
Uccelli A, Moretta L, Pistoia V (2008) Mesenchymal stem cells in health and disease. Nat Rev Immunol 8: 726-736.
