

Journal of Genetics and Genome Research
Replacing CFTR Sanger Sequencing in the Clinical Lab with a Reliable, Targeted Next-Generation Sequencing Assay
Shela Lee, Joy Radecki, Hsiao-Mei Lu and Aaron M. Elliott*
Ambry Genetics, USA
*Corresponding author:
Aaron M. Elliott, Ambry Genetics, 15 Argonaut, Aliso Viejo, California, USA, E-mail: aelliott@ambrygen.com
J Genet Genome Res, JGGR-1-004, (Volume 1, Issue 1), Research Article; ISSN: 2378-3648
Received: September 04, 2014 | Accepted: October 01, 2014 | Published: October 05, 2014
Citation: Lee S, Radecki J, Lu HM, Elliott AM (2014) Replacing CFTR Sanger Sequencing in the Clinical Lab with a Reliable, Targeted Next-Generation Sequencing Assay. J Genet Genome Res 1:004. 10.23937/2378-3648/1410004
Copyright: © 2014 Lee S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
The clinical implementation of new target enrichment methods and next-generation sequencing (NGS) technology has rapidly transformed genetic testing. Diagnostic labs can now offer a wide variety of large comprehensive multi-gene panels or even full exome sequencing to help clinicians diagnose and treat patients. The unmatched sensitivity, accuracy and throughput of NGS compared to traditional Sanger sequencing make it an ideal technology not only for panels but also high volume single gene assays. Here we describe the validation and performance of an NGS based assay for sequencing the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. The custom designed assay utilizes TruSeq Custom Amplicon (TSCA) target enrichment and modified bioinformatics pipeline to identify different classes of mutations, including small deletions and insertions. Validation of the test with 151 previously characterized CFTR variants resulted in 100% accuracy. Test specificity of 99.99% was determined by analyzing Sanger sequencing confirmation data from the first 2,000 samples. In addition, the assay was able to detect variants missed by previous testing due to allele-dropout. The transition of the CFTR sequencing assay from Sanger sequencing to a custom NGS based test has not only increased the sensitivity and reliability of the assay but also cut the turn-around-time in half, allowing clinicians to diagnose and make treatment decisions quicker.
Keywords
Sanger sequencing; Next generation sequencing; NGS; Diagnostics; Genetic testing
Introduction
For the last 30 years, Sanger dideoxy terminator sequencing has been considered by many to be the gold-standard for decoding DNA [1]. Most diagnostic laboratories routinely use Sanger sequencing for single and multi-gene variant detection due to its robustness, high accuracy and ease of clinical set-up. However, Sanger sequencing has several limitations that hinder a modern clinical diagnostic lab. First, directly sequencing PCR products by Sanger is hard to scale and not feasible for screening large sets of genes or even single genes in high volume, due to the costs and resources needed [2]. Second, Sanger sequencing is limited in its sensitivity and ability to analyze allele frequencies [3]. Finally, analyzing Sanger sequencing data in a CLIA/CAP accredited lab is extremely time consuming due to the need of a licensed Clinical Laboratory Scientist (CLS) to manually analyzethesequencing chromatograms for a given gene.
Recent advancements in target enrichment and Next-Generation Sequencing (NGS) technologies have made it possible to generate large amounts of data quickly and cost effectively, eliminating the throughput and resource constraints seen with Sanger sequencing. Moreover, data is processed by bioinformatics scientists and results generated without the need for manual CLS review of every base pair. There have been numerous reports in the literature about the benefits of using various target enrichment methods and NGS to sequence large gene panels and exomes in clinical diagnostics [4-6]. In these cases, highly multi-plexed target enrichment techniques and NGS are the only methods possibleto sequence a multitude of genes simultaneously and typically have Turn-Around-Times (TAT) in the range of weeks to months. Currently many labs continue to use Sanger sequencing for single and small gene panels due to throughput and costs with a standard TAT averaging 3-4 weeks. Importantly, target enrichment and NGS is only cost-effective and efficient if a laboratory receives the appropriate sample volume. For low volume assays to be cost effective on an NGS platform they either need to be batched which increases TAT or multiplexed with other gene assays. Moreover, the experience needed to process samples in a diagnostic setting usinghigh-throughput target-enrichment and NGS cannot be overstated. However, for those diagnostic laboratories with the experience and volume required, transitioning from Sanger to NGS can result in increased accuracy, cost savings and faster TAT. Delivering results to the patient in a timely manner is extremely important as many times diagnosis alters medical management and in some instances treatment or surgery could be pending the results.
Here we describe the implementation of a target enrichment and NGS workflow for sequencing the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene. Ambry Genetics was the first laboratory to offer clinical grade sequencing of the full CFTR gene over a decade ago. With over 35,000 full gene CFTR samples sequenced to date, we have the most extensive database and mutant repository in the world. Utilizing these resources, as well as our vast clinical target enrichment andNGS experience, we detail the transition of our PCR amplicon based Sanger CFTR assay over to a custom designed TruSeq Custom Amplicon (TSCA) target enrichment and NGS assay. This approach coupled with our custom bioinformatics pipeline has increased our test accuracy and sensitivity while decreasing our test TAT from 14-28 days to 5-13 days, allowing clinicians to diagnose and make treatment decisions quicker.
Materials and Methods
DNA Samples
The CFTR NGS assay was conducted on 43 previously characterized, archived genomic DNA samples and an additional 2,000 clinical samples sent in for testing.All individuals used for testing provided written consent. All data was de-identified prior to analysis. At least 6~7μg of genomic DNA was extracted from whole blood or saliva using the QiaSymphony instrument (Qiagen) according to the manufacturer's instructions. Isolated DNA was quantified using a NanoDrop UV spectrophotometer (Thermo Scientific).
TruSeq Custom Amplicon library preparation
Using Illumina Design Studio, a pool of custom oligonucleotides were designed to fully target and capture all CFTR exons, the 5'UTR region, two deep intronic regions and at least 20 base pairs flanking exons. For each sample =250 ng of genomic DNA was used to generate TSCA libraries using Illumina's TruSeq Custom Amplicon Kit (Cat # FC-130-1001) according to the Illumina protocol (#15027983 Rev.A). In brief, a template library is generated by hybridization of CFTR oligonucleotide probes to unfragmented genomic DNA, followed by extension and ligation resulting in DNA templates consisting of regions of interest flanked by universal primer sequences. Indices and sequencing adapters are thenattached to the template by PCR, purified, quantified and normalized to 2nM. Samples are then pooled, and sequenced on the Illumina MiSeq. Base calling and annotation were performed using Ambry's custom built bioinformatics pipeline. Illumina Design Studio does not allow custom sequence input and will not accommodate breakpoint probe design for large deletions. Secondary amplicons, "add-ons", were developed in house to achieve full coverage of the CFTR targeted regions and common large deletion breakpoints.
CFTR Add-On Library Preparation
Add-On primers were designed in Vector VNTI Advance v11.5.1 (Invitrogen) with TSCA adapter sequences attached. For PCR amplification, 50 ng (50ng/μl) of genomic DNA was added to 5 μl HotStarTaq Master Mix (Qiagen), 1 μl of tailed primers (2.5 μM and .5 μlM), 1 μl of i5 2.5 μM Primer Index, 1 μl of i7 2.5 μM Primer index and 1μl of nuclease-free water. PCR amplification was performed in a Bio-Rad MyCycler (Bio-Rad) with the following conditions: 95°C for 15 min, followed by a program of 94°C for 30 s, 61°C for 30 s, and 72°C for 45s for 35 cycles and ending with a 10 min extension at 72°C.PCR products were purified usingAMPure XP beads (Beckman Coulter) according to the manufacturer's instructionsand pooled into a single tube per amplicon. Amplicon libraries were quantified using the Bioanalyzer DNA 1000 kit (Agilent Technologies), and normalized to combine with the associated TSCA library.
Sequencing and analysis parameters
TSCA libraries were sequenced on the Illumina MiSeq platform using sequencing-by-synthesis technology using 150 base paired-end reads. Sequence read data were analyzed using the Ambry custom bioinformatics pipeline for CFTR TSCA. Initial data processing and base calling, including extraction of cluster intensities, was done locally on the instrument control PC using MCS 1.2.3. and RTA 1.14.23. Sequence quality filtering script was executed in the Illumina CASAVA software V.1.8.2. The MiSeq Reporter V.1.3.17 (Illumina) software was used to detect SNPs, variants and indels, and to generate coverage and no coverage reports. Reads were accurately aligned to a reference sequence and base-calls that differed from the reference were evaluated to identify possible biases. The variant calling filters used were: quality score = 20 and coverage = 10x.These two values were determined empirically to ensure a mutation detection rate of 100% with minimal false-positive calls.Sanger sequencing confirmation was performed for all sequence alterations within the analytical range of the test.
Results and Discussion
CFTR NGS Assay work flow
The development ofa high volume diagnostic sequencing test is a more challenging and time consuming undertaking compared to implementing a research grade assay. When designing probes or primers for target enrichment, high frequency SNPs (> 1%) must be avoided to limit allele drop-out. In addition, the assay must be able to identify all known pathogenic variants within the reportable range of the test. Any variant or region which cannot be reliably detected by NGS needs to be sequenced by Sanger sequencing toprovide adequate coverage. Finally, the assay work-flow needs to be suitable for high-throughput andmeet the requirements of a CLIA/CAP accredited lab. Due to all these factors, most diagnostic assays need to be custom developed and cannot be directly purchased as a catalog product from a vendor.
From a technical standpoint, the CFTR NGS assay provides several advantages compared to the traditional Sanger sequencing test and other CFTR NGS approaches which enable a faster diagnostic TAT. The CFTR NGS assay is comprised of 61 amplicons in a single reaction compared to 33 primer pairs and 66 reactions (forward read and reverse read) for Sanger sequencing. In addition, the workflow is highly automated with the majority of time saved in not having to analyze Sanger chromatograms for every region sequenced. The integrated dual indexing during PCR enables up to 96 samples (including positive and negative control) per NGS run on the MiSeq (Illumina), drastically shortening the time and expense compared to Sanger sequencing (Figure 1).
 Figure 1: Timeline for reporting 94 CFTR Samples using Sanger
sequencing vs a targeted TSCA NGS workflow.
View Figure 1
Figure 1: Timeline for reporting 94 CFTR Samples using Sanger
sequencing vs a targeted TSCA NGS workflow.
View Figure 1
CFTR NGS assay design
The custom designed CFTR NGS sequencing test includes comprehensive analysis of all 27 coding exons and at least 20 bp into the flanking intronic sequences and 5' and 3' untranslated regions. The assay was also developed to target the Poly-Thymidine (poly-T) and poly-thymidine-guanine (poly-TG) tracts of exon 10 [7,8], identify the c.1679+1634 A>G mutation in intron 12, and the c.3717+12191C>T mutation in intron 22.The assay is designed to detect nucleotide substitutions, small deletions, small insertions (including small repeat expansions) and small indels. To identify large deletions and duplications beyond the capability of NGS detection, Multiplex Ligation-Dependent Probe Amplification (MLPA) is done concurrently for all samples.
When transitioning a diagnostic assay to a new technology (Sanger to NGS), it is extremely important to have a good understanding of the variants characterized previously. For example, an assay could have limitations in detecting medium size deletions. These are deletions that are too big to detect by target enrichment and NGS but too small to pick up with MLPA or a microarray. By analyzing the genomic coordinates of the probe design we were able to determine if the design could detect the mutations in our extensive CFTR database. In the TSCA design there was a probe placed over a known 84 base pair deletion in exon 14 (c.1817_1900del84) which would cause the mutant allele to drop out, resulting in the sample appearing wild-type (false negative). Unfortunately, the Illumina design software does not allow the user flexibility to determine where probes are placed nor does it enable the user to detect breakpoints. To resolve this issue, add-on break point PCR primers were designed to detect the del84 variant and the amplicons combined with the TSCA final library for NGS sequencing.
Bioinformatics and software employed for data analysis in diagnostic assays introduces arguably the most variability in accuracy and sensitivity between different NGS tests. It is imperative that the bioinformatics pipeline be specifically tailored for the gene being tested and account for the methods used for enrichment and sequencing. During development of the CFTR NGS bioinformatics pipeline, we discovered the primer trimming function in the standard Illumina MiSeq Reporter resulted in unreliable variant calling in regions neighboring primer sequences. For example, a one base pair deletion close to the trimmed off primer sequence is problematic for the variant caller as it cannot distinguish whether it is a true deletion or a sequencing error in the beginning of the read. However, a one base pair deletion can easily be detected by aligning untrimmed primer sequencing reads because the one base pair gap sits close to the center of the read. Therefore, in order to increase the accuracy and sensitivity of calls near primer binding sites, we customized our bioinformatics pipeline to cross check variants on and near primer regions using both trimmed and untrimmed reads.
Analytical validity of CFTR NGS assay
To determine the accuracy and sensitivity of the CFTR assay, a cohort of 43 previously Sanger sequenced DNA and saliva samples were selected which represented a variety of different classes of variants including small insertions and deletions. The average read depth across all samples was extremely high at 2,900X. The46 samples harbored 151 previously detected germline variants, which were all correctly identified using the NGS CFTR assay, resulting in 100% sensitivity (Table 1) (Figure 2). Included in the accuracy samples was the c.1817_1900del84 variant which is detected using the add-on breakpoint primers.There were no false positives identified in the accuracy samples. However, the false positive rate of an assay cannot typically be well defined until hundreds or thousands of samples are processed. Therefore, to better determine the false positive profile of the assay, after clinically launching the test those variants detected by NGS were Sanger confirmed. Following the analysis of 2,000 patient samples submitted for CFTR full gene sequencing using the custom TSCA NGS assay there were 115 homozygous variants and 1,020 heterozygous variants confirmed. In the first 2,000 samples we detected 40 false positives (Figure 3). All false positive calls were at a read ratio below 20% or very low coverage. The low false positive rate of the assay, which reduces the need for time consuming Sanger sequencing confirmation, is crucial to meet the 5-13 day TAT.
![]()
Table 1: Accuracy variant detection on previously characterized samples.
View Table 1
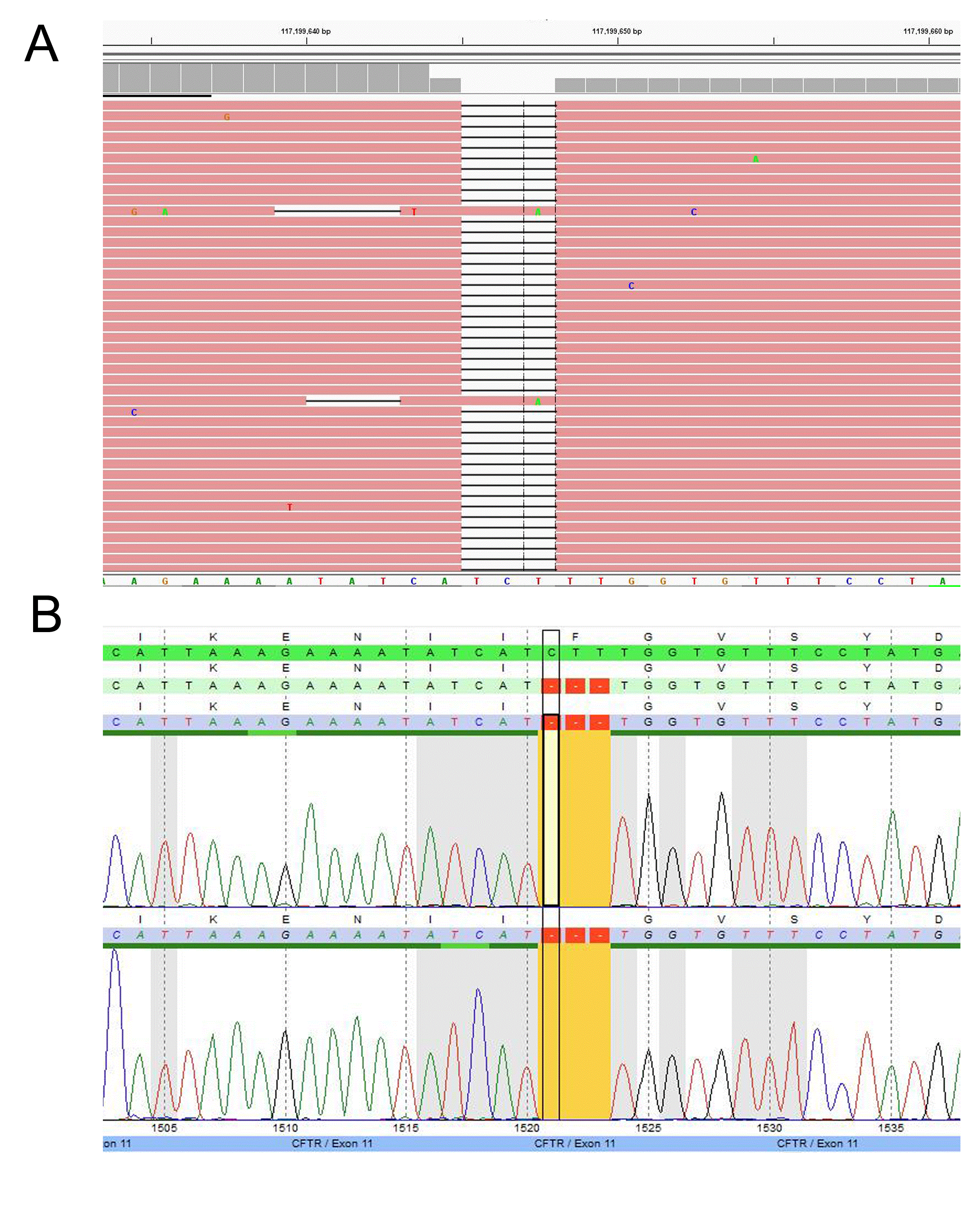 Figure 2: Example of a CFTR mutation detected by the TSCA NGS assay
(A) Homozygous small deletion at c.1521_1523delCTT (p.F508del) identified by NGS and (B) Sanger confirmed.
View Figure 2
Figure 2: Example of a CFTR mutation detected by the TSCA NGS assay
(A) Homozygous small deletion at c.1521_1523delCTT (p.F508del) identified by NGS and (B) Sanger confirmed.
View Figure 2
A major concern of diagnostic sequencing is allele drop-out which often produces false negatives. This occurs when a variant located underneath a primer binding site interrupts hybridization resulting in amplicon drop-out [9,10]. If a variant is located on the same allele and within the affected amplicon, the variant will go undetected. Allele drop-out is a major concern in primer based target enrichment, such as that use in Sanger sequencing, as one variant underneath a primer is typically sufficient to cause drop-out. In our TSCA CFTR design we tested several samples with known variants underneath the probe binding sites and did not observe a significant effect on allele bias. Notably, the new design has detected variants in several samples that were previously missed by Sanger sequencing. For example, the TSCA NGS CFTR assay detected a sample with a c.3154 T>G heterozygous variant, which had previously been identified as homozygous wild-type by Sanger sequencing (Figure 4). Upon further inspection, a variant was detected underneath the PCR primer binding siteresulting in allele-drop out. Typically, when utilizing Sanger sequencing, allele drop-out is only detected when the wild-type allele is affected, leaving a suspicious homozygous mutation call [10].
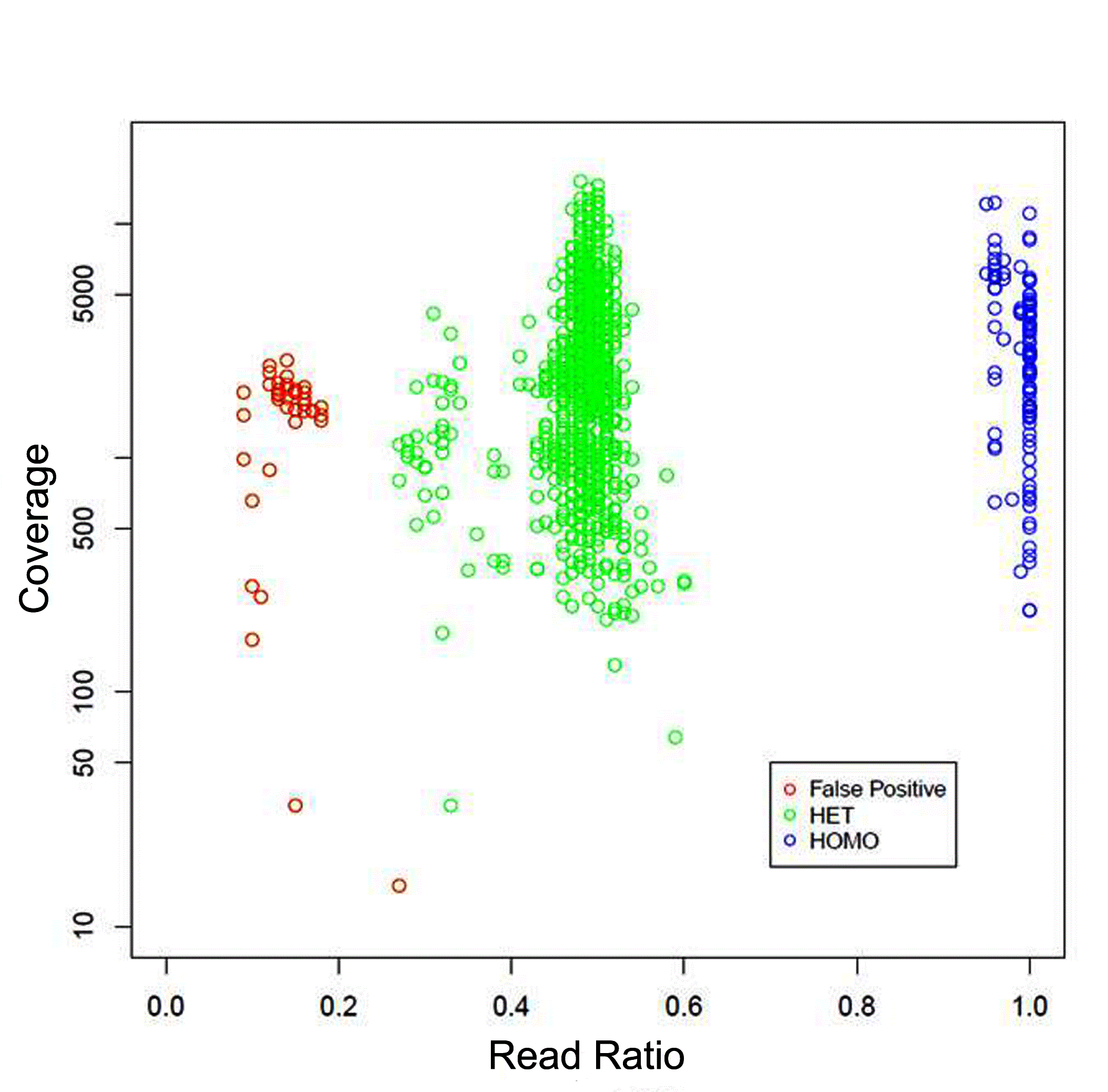 Figure 3: Variant read ratio vs. read coverage in first 2,000 samples
The false positive profile of the NGS CFTR assay was determined by
plotting the sequencing coverage against the variant read ratios using
Sanger sequencing confirmed NGS variants. Red circle, Sanger cleared
false positive. Green circle, Sanger confirmed heterozygous NGS variant.
Blue circle, Sanger confirmed homozygous NGS variant.
View Figure 3
Figure 3: Variant read ratio vs. read coverage in first 2,000 samples
The false positive profile of the NGS CFTR assay was determined by
plotting the sequencing coverage against the variant read ratios using
Sanger sequencing confirmed NGS variants. Red circle, Sanger cleared
false positive. Green circle, Sanger confirmed heterozygous NGS variant.
Blue circle, Sanger confirmed homozygous NGS variant.
View Figure 3
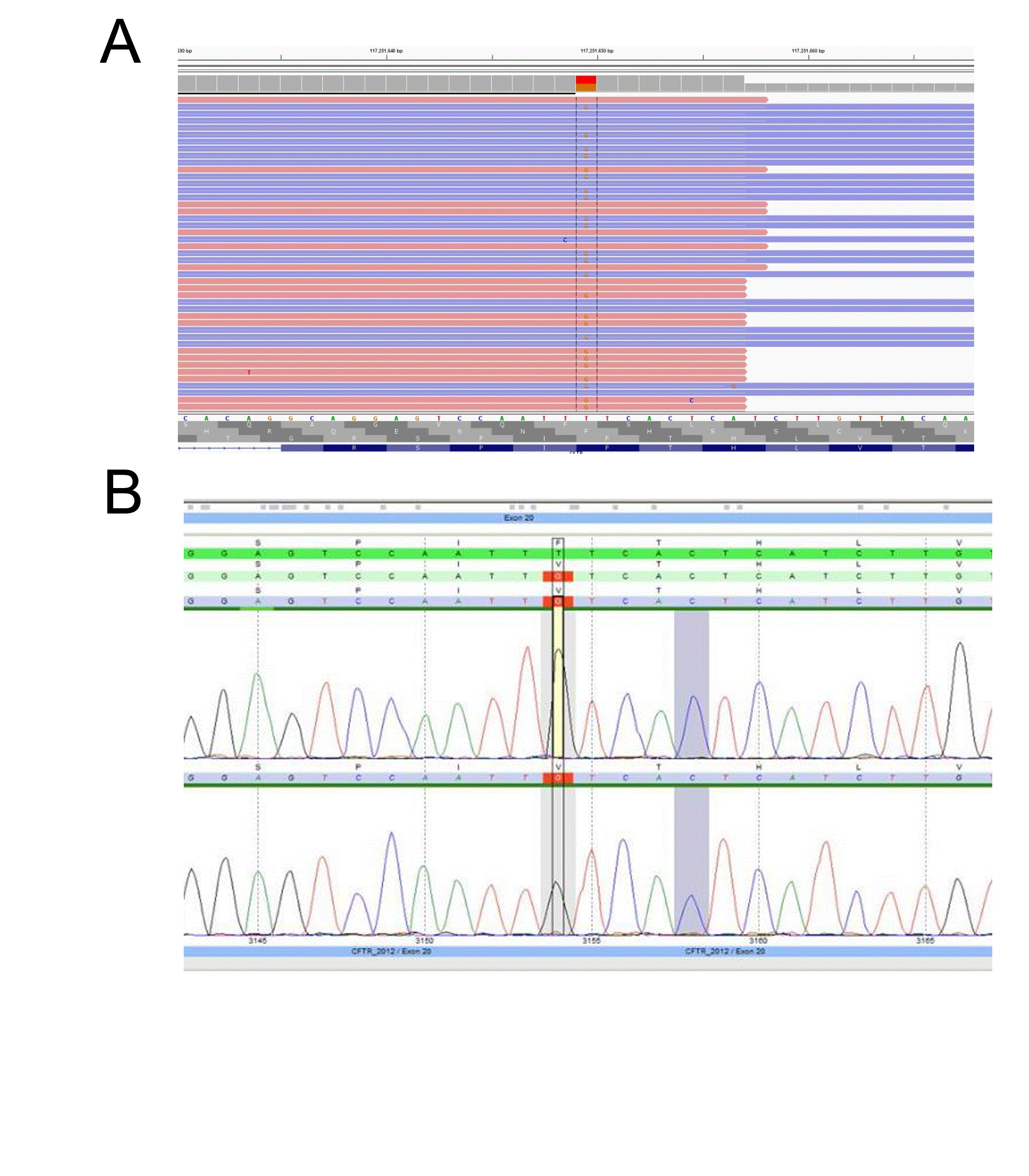 Figure 4: CFTR NGS assay accurately detects variants missed by Sanger sequencing due to allele-dropout
(A)Sample with a c.3154 T>G heterozygous variant detected by NGS and (B) missed by Sanger sequencing.
View Figure 4
Figure 4: CFTR NGS assay accurately detects variants missed by Sanger sequencing due to allele-dropout
(A)Sample with a c.3154 T>G heterozygous variant detected by NGS and (B) missed by Sanger sequencing.
View Figure 4
To determine the precision and reproducibility of the assay 16 previously characterized blood and saliva samples harboring a total of 53 variants were processed through the entire work flow three times and concordance between runs determined. There was no significant variability detected between repeats as all 53 variants were detected in each run resulting in 100% reproducibility.
Other NGS CFTR assays
There have been numerous publications detailing the design and validation of NGS based CFTR assays [11-14]. Importantly, there are significant differences between the CFTR sequencing test described here, which is designed to process clinical samples in high volume, and other published assays. When implementing a diagnostic test into clinical practice, the accuracy and specificity of the assay cannot be compromised for throughput and speed. Recently, AbouTayoun et al. described a comprehensive CFTR NGS assay utilizing Ion Torrent semiconductor sequencing technology [11]. In concordance with a 2012 published study detailing CFTR Ion Torrent sequencing, the authors observed false positive calls in homopolymer stretches, a common problem with flow based chemistry[12]. In our experience, these can be accounted for bioinformatically, however it is generally at the expense of missing true calls in the region. Ultimately, a lab would need to Sanger sequence these homopolymer regions to be confident in making the correct call, costing time and money.
In addition to accuracy and specificity, diagnostic labs processing large numbers of samples need an assay designed for high throughput and fast TAT. The CFTR NGS assay described by Trujillano et al. utilized NimbleGenSeqCap probe based target enrichment with IlluminaHiSeq sequencing [13]. The complete CFTR gene was tiled with probes to accurately detect not only small base pair variations but also gross deletions and duplications. However, the assay workflow is long and cumbersome. First, the starting material required is over 1μg, which could be a significant limitation for some sample types such as blood spots. In addition, there are several time consuming steps such as sonication, library preparation and a 48-72 hour hybridization, which significantly increases the time required to process samples. Moreover, only 24 samples were multiplexed per lane of sequencing reducing the throughput.Therefore, with the described workflow it would be nearly impossible to generate a diagnostic report within the proposed low end TAT of 5 days.
The IlluminaMiseqDx CFTR NGS assay is an FDA-cleared in vitro diagnostic (IVD) system [14]. The assay uses the same workflow and TSCA technology described here. However, there are several disadvantageswhen using a commercially available kit that diagnostic labs need to be aware of. Users don't have the flexibility to alter the design to detect more complicated mutations or those published in the future as being causative. This is a big disadvantage of using any FDA approved test for target enrichment and gene sequencing, as most vendors are extremely reluctant to alter a design after achieving regulatory approval. For example, there is no indication the MiSeqDx CFTR assay can detect the causative c.1817_1900del84 mutation. With the rapid advances in technology and variant discovery, this is a major topic that will need to be addressed if genetic testing is to be regulated by the FDA. In addition, users pay a premium for the MiSeqDx instrumentation and reagents due to its regulatory status. For smaller labs without the experience or resources to design their own assays, the MiseqDx CFTR test is a valuable resource. However, for larger, more experienced labs the flexibility and clinical accuracy of an assay is generally improved when designed and validated in house.
Here we describe the transition of CFTR, a high volume single gene diagnostic assay, from Sanger sequencing to NGS. The test was able to detect all previously characterized variations and identify other calls missed by Sanger sequencing. The reliability, low false positive rate and streamlined workflow will allow clinicians to receive results in a timely manner to aid in diagnosis and treatment decisions.
References
-
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74: 5463-5467.
-
Oetting WS (2010) Impact of next generation sequencing: the 2009 Human Genome Variation Society Scientific Meeting. Hum Mutat 31: 500-503.
-
Chin EL, da Silva C, Hegde M (2013) Assessment of clinical analytical sensitivity and specificity of next-generation sequencing for detection of simple and complex mutations. BMC Genet 14: 6.
-
Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM, et al. (2012) ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn 14: 357-366.
-
Chong HK, Wang T, Lu HM, Seidler S, Lu H, et al. (2014) The validation and clinical implementation of BRCAplus: a comprehensive high-risk breast cancer diagnostic assay. PLoS One 9: e97408.
-
Helsmoortel C, Vandeweyer G, Ordoukhanian P, Van Nieuwerburgh F, Van der Aa N, et al. (2014) Challenges and opportunities in the investigation of unexplained intellectual disability using family based whole exome sequencing. Clin Genet .
-
Chillón M, Casals T, Mercier B, Bassas L, Lissens W, et al. (1995) Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 332: 1475-1480.
-
Groman JD, Hefferon TW, Casals T, Bassas L, Estivill X, et al. (2004) Variation in a repeat sequence determines whether a common variant of the cystic fibrosis transmembrane conductance regulator gene is pathogenic or benign. Am J Hum Genet 74: 176-179.
-
Lam CW, Mak CM (2006) Allele dropout in PCR-based diagnosis of Wilson disease: mechanisms and solutions. Clin Chem 52: 517-520.
-
Landsverk ML, Douglas GV, Tang S, Zhang VW, Wang GL, et al. (2012) Diagnostic approaches to apparent homozygosity. Genet Med 14: 877-882.
-
Abou Tayoun AN, Tunkey CD, Pugh TJ, Ross T, Shah M, et al. (2013) A comprehensive assay for CFTR mutational analysis using next-generation sequencing. Clin Chem 59: 1481-1488.
-
Elliott AM, Radecki J, Moghis B, Li X, Kammesheidt A (2012) Rapid detection of the ACMG/ACOG-recommended 23 CFTR disease-causing mutations using ion torrent semiconductor sequencing. J Biomol Tech 23: 24-30.
-
Trujillano D, Ramos MD, González J, Tornador C, Sotillo F, et al. (2013) Next generation diagnostics of cystic fibrosis and CFTR-related disorders by targeted multiplex high-coverage resequencing of CFTR. J Med Genet 50: 455-462.
-
Grosu DS, Hague L, Chelliserry M, Kruglyak KM, Lenta R, et al. (2014) Clinical investigational studies for validation of a next-generation sequencing in vitro diagnostic device for cystic fibrosis testing. Expert Rev Mol Diagn 14: 605-622.
