

Journal of Hypertension and Management
The Degree of Apoptosis in the Cytotrophoblast Regulates Amyloidosis Fate in the ER Stress of the Syncytiotrophoblast: Morphological Evidence
C Bosco1*, M Parra-Cordero2 and E Díaz1
1Institute of Biomedical Sciences, University of Chile, Chile
2Department of Obstetrics and Gynecology, University of Chile, Chile
*Corresponding author:
C Bosco, Anatomy and Developmental Biology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago, Chile, E-mail: cbosco@med.uchile.cl
J Hypertens Manag, JHM-2-022, (Volume 2, Issue 2), Original Article; ISSN: 2474-3690
Received: October 31, 2016 | Accepted: December 19, 2016 | Published: December 21, 2016
Citation: Bosco C, Parra-Cordero M, Díaz E (2016) The Degree of Apoptosis in the Cytotrophoblast Regulates Amyloidosis Fate in the ER Stress of the Syncytiotrophoblast: Morphological Evidence. J Hypertens Manag 2:022. 10.23937/2474-3690/1510022
Copyright: © 2016 Bosco C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Recently preeclampsia (PE) has been linked throught molecular evidence to endoplasmic reticulum (ER) stress in the placenta. In this organ ER stress is generated due to placental oxidative stress as a consequence of malperfusion secondary to deficient spiral artery remodeling. Immunohistochemistry and ultrastructural analysis were used to study tissue specimens from 14 term placentas: 4 normal, 4 PE, 3 intrauterine growth restriction (IUGR), and 3 PE+IUGR. Antihuman Amyloid A mouse monoclonal antibody (AA) and antihuman mouse monoclonal antibody to active caspase 3 were used in the immunohistochemistry study. Ultrastructural analysis was also performed by transmission electron microscopy. We found that cytotrophoblast (CT) apoptosis was increased in presence of amyloidosis. In IUGR placentas, amyloid deposits were located on the trophoblast basal lamina and were not observed into the cytoplasm of the syncytiotrophoblast (ST); in PE amyloidosis was evidenced throughout the cytoplasm of CT and ST cells, with expulsion of ST microparticles into the maternal blood. In PE+IUGR condition, amyloid deposits were observed similar to PE but reaching as far as the fetal endothelium. Additionally we found presence of activated caspase 3 in the CT and ST of PE placentas. We propose that the degree of CT apoptosis regulates the fate of the amyloid deposits produced by ER stress in the ST. The morphological and immunohistochemical evidence provided in this work support the inclusion of PE in the group of conformational or protein misfolding diseases.
Keywords
Placenta, Preeclampsia, Trophoblast, IUGR, ER stress
Introduction
PE is a pregnancy pathology affecting 3-5% of pregnancies, associated with both maternal and fetal morbidity and mortality [1]. Its etiology is unknown, although it has been postulated that its cause is related to the placenta [2] due to the fact that the symptoms disappear once the organ is expelled [3]. So far, there is no treatment for PE. The mother develops proteinuria, edema and experiences a rise in blood pressure [4], affecting normal fetal development. Currently, the only possible treatment for this disease is early delivery with the consequent birth of premature infants [5].
To date, the studies on PE have shown a relationship with placental malperfusion, secondary to deficient spiral artery conversion into low resistance high-flow vessels in the endometrium [6], leading to an increase in the formation of reactive oxygen species [3,7,8] that finally leads to the development of oxidative stress associated with endothelial dysfunction, abnormal levels of nitric oxide (NO), aberrant angiogenesis and increased placental apoptosis [9-11]. Additionally, Hung, et al. [9] proposed that the cause of the oxidative stress in the placenta is an injury of the ischemia-reperfusion type. Recent studies suggest that PE could be triggered by disorders in the folding of proteins in the endoplasmic reticulum (ER) of the syncytiotrophoblast (ST), which results in amyloid deposits in this organelle [12-14]. Due to the fact that several proteins share the property of conforming as antiparallel beta-sheets, and forming insoluble amyloid fibrils that deposit in the organ/tissue causing systemic amyloidosis, PE is considered to be one of the so called "conformational diseases" [12-14].
It is known that in order to fulfill its physiological functions, a protein must be correctly folded in its secondary structure, given by the folding determined by aminoacid residues in close proximity in the polypeptide chain, thus facilitating the formation of hydrogen bonds between the nearby carbonyl (-CO-) and amino (-NH-) groups. There are two types of protein secondary structure, the alpha helix configuration and the beta sheet folding. The latter is stabilized by intermolecular interactions, in the tertiary structure, that eventually constitutes oligomers, proto-fibrils and fibrils, which accumulate forming amyloid deposits in the extracellular matrix and/or in the cytoplasm, thereby affecting normal tissue function [15-17]. An example of this is the toxic deposit of amyloid -beta A (Aβ) in Alzheimer's disease [14] and the amyloid polypeptide (IAPP) deposits in beta cells of the pancreatic islets in Type 2 diabetes, to name a few of these pathologies [18]. Protein misfolding generates insoluble, proteolysis resistant structures, with the subsequent loss of its physiological function or the generation of toxic compounds that cause pathological events.
The ER is the organelle where secreted or membrane proteins are synthesized. They are secreted once the processes of folding, glycosylation and formation of bi-sulfide bonds are completed. Normal folding requires the nascent protein to be maintained in the ER lumen, where it is attached to a Ca+2 -dependent chaperone [19]. For this reason, the lumen of the ER contains a large amount of Ca+2 ions which accumulates at expense of an active transport process regulated by Ca2+ -ATPase. In addition, the ER lumen must also provide an oxidative environment in order to promote the formation of disulfide bonds, [14,20-23]. The accumulation of misfolded protein in the ER lumen has been defined as 'ER Stress' [20,24,25]. Such stress is caused by dysregulation of the ER homeostasis as a result of extracellular stimuli, which in turn results in an accumulation of these misfolded proteins inside the ER, thus causing blockade of the protein migratory way to the Golgi apparatus. As a consequence, a cellular response known as Unfolded Protein Response (UPR), widely described in the literature [23,25-27] is activated. The UPR has two main objectives: (i) First, to regain normal cell function by stopping protein translation and activating signaling pathways that allow an increase in the production of molecular chaperones involved in the process of protein folding. So, the initial aim of the UPR is to resolve the defect and to restore ER homeostasis; (ii) Secondly, if the cell does not reach the first goal in a certain period of time, or if the problem persists, the UPR triggers cellular apoptosis [28-32]. PKR-like endoplasmic reticulum kinase (PERK) is an ER protein, usually inactive, whose function is to sense protein misfolding. However, before the accumulation of misfolded proteins, PERK is activated by dimerization and autophosphorylation [27]. The activation of PERK results in phosphorylation of the alpha subunit of the eukaryotic initiation factor 2 (eIF2α), which quickly blocks protein translation, thus reducing the protein burden within the ER.
It has been shown that the activation of the UPR in placentas with IUGR and IUGR+PE triggers an increased phosphorylation of eIF2α in both pathological conditions, compared to normal controls, reaching the greatest extent in IUGR+PE placentas [23,31]. These authors concluded that although ER stress and the activation of the UPR was evident in IUGR and PE+IUGR placentas, the response was far more extensive in the latter, which was associated with the increased apoptosis observed in IUGR+PE trophoblast and the consequent release of microparticles to the blood hematic chamber [31,33]. Additionally, it has been shown in vitro by Yung, et al. [24] + that ER stress exacerbates the ischemia/reperfusion induced by apoptosis through attenuation of the Akt protein synthesis in human choriocarcinoma JEG-3 cells used as a model for placental trophoblast.
Substantiated on the body of evidence provided, the aim of this study was to analyze placentas from women suffering from IUGR, PE and PE+IUGR versus control placentas by two lines of approach: i) transmittion electron microscopy (TEM) level, ii) immunohistochemistry using mouse monoclonal antiamyloid A (AA) antibody and mouse monoclonal antibody to active-Caspase 3, in order to evidence possible areas of accumulation of amyloid deposits in pathological placentas and the degree of apoptosis in both the CT and the ST.
Material and Methods
This study was conducted on a group composed of 14 pregnant women enrolled for prenatal care in the Department of Obstetrics and Gynecology, University of Chile Clinical Hospital. Four groups, composed of i) 4 PE, ii) 3 IUGR, iii) 3 IUGR+PE and iv) 4 control women were included, in accordance with our previous classification [34]. Briefly, the criteria for the inclusion in the PE group required the participants to be healthy prior to pregnancy, with no history of hypertension, diabetes or renal dysfunction. PE parameters have been previously defined [35] based on the classification of the American College of Obstetrics and Gynecology. Hypertension and proteinuria levels were required to return to normal values after delivery. The criteria for the inclusion in the control group was normal pregnancy, defined as a pregnancy in which the mother had normal blood pressure, absence of proteinuria, and no previous medical or pregnancy complications. Exclusion criteria for the control group included: chronic diseases such as diabetes mellitus, known chronic hypertension, major fetal abnormalities, placental tumor, intrauterine infection, twin pregnancies, obstetric pathologies other than spontaneous preterm delivery and maternal disease, malignant neoplasia, dyslipidemia, obesity or others pathologies known to cause alterations in the biochemical parameters of oxidative stress and/or endothelial dysfunction. IUGR was defined as neonatal weight below 10th percentile for the Chilean population [36]. Clinical data of the participants in the present study are shown in table 1.
![]()
Table 1: Clinical characteristics of the participant patients.
View Table 1
Placentas were obtained immediately after delivery. Samples from 4 human term normal placentas, 4 PE, 3 IUGR and 3 IUGR+PE placentas were routinely processed for immunohistochemical assay as previously reported [34]. Briefly, for the amyloid determination, mouse monoclonal Anti- human Amyloid A (AA), 1:100 (v/v) (DAKO, Code N° 0759), and mouse monoclonal anti-human active Caspase 3 (CPP 32), 1:50 (v/v), (Novocastra JHM62) were separately applied to histological sections for 30 min at 37°C. Immunostaining was revealed using the horseradish peroxidase-labelled streptavidin biotin kit (DAKO, USA), according to the manufacturer's directions, using 3, 3'diaminobenzidine (DAB) as chromogen. In negative controls the antibody was omitted. In addition, small tissue samples from the same placentas were routinely processed for TEM as previously described [37]. Women included in the study were required to sign an informed consent form and all procedures were performed according to protocols approved by the local ethic committees of the Faculty of Medicine of the University of Chile and the Clinical Hospital.
Results
Control placentas
Control samples treated with immunohistochemistry for AA antibody, showed no amyloid expression in ST nor in CT cells (Figure 1A); this was only evidenced in stromal areas, where the amyloid was intermingled with fibrinoid deposits (data not shown). Control samples treated with anti-active Caspase 3 antibody showed positive reaction only in the ST (Figure 2A).
.
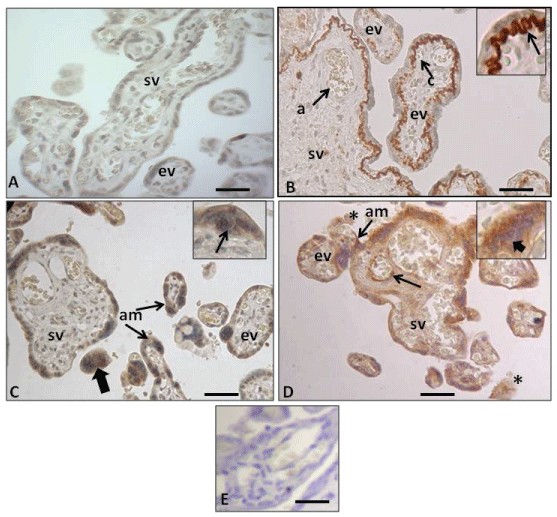
Figure 1: Immunohistochemistry of human placental barrier. (A) Expression of the anti-Amyloid A mouse monoclonal antibody in control placenta, displaying no immunohistochemical reaction in the trophoblast epithelium (CT and ST), the endothelium and stroma. Terminal stem villi (sv) and exchange villi (ev); (B) Expression of the same antibody in IURG placenta displaying positive immunohistochemical mark. Note that in this case only the basal lamina of the trophoblast of the stem villi evidence a very intense reaction (arrow in inset). Stem villi (sv) displaying an arterie (a) and exchange villi (ev) showing a blood capillar (c); (C) Expression of the same antibody in PE placenta, displaying positive mark in the ST cytoplasm (am) and in the CT cytoplasm (arrow in inset), and the syncytial nots (thick arrow); (D) Expression of the same antibody in PE-IURG placenta, displaying an apoptotic CT (thick arrow in inset) and intense amyloid deposits (am) in the ST of stem villi (sv) and exchange villi (ev) in cellular debris in the maternal space (asteriks) and in the endothelium of the fetal vessel (arrow) of a stem villi (sv); (E) Placenta from the normotensive pregnancy group showing negative control anti Amyloid A staining. The negative control was incubated with no primary antibody.
Antibody working dilution 1:100. Calibration bars 50 μm.
View Figure 1
.
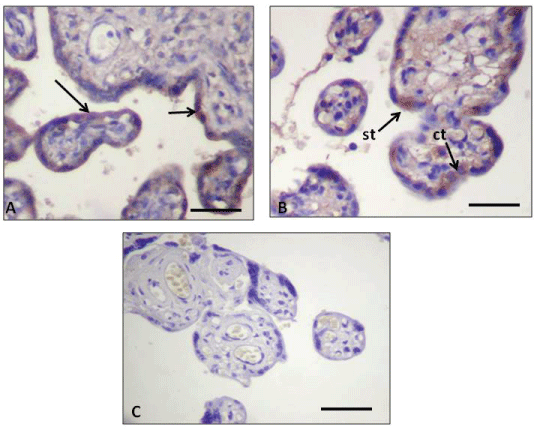
Figure 2: Immunohistochemistry of human placental barrier. (A) Expression of the anti-active Caspase 3 antibody in control placenta displaying immunohistochemical reaction in the ST (arrows); (B) Expression of the same antibody in PE placenta displaying a positive immunohistochemical mark in two CT cells (ct) and in the ST (st); (C) Placenta from the PE pregnancy group showing negative control for active-Caspase 3 staining. The negative control was incubated with no primary antibody in a PE sample.
Antibody working dilution 1:50. Calibration bars 50 μm.
View Figure 2
These results are consistent with the aspect of the ER observed at TEM level, that our control samples evidenced normal cisternae in the ST of the exchange villi (Figure 3A), where the presence of an underlying CT, displaying normal cytological features was also observed.
.
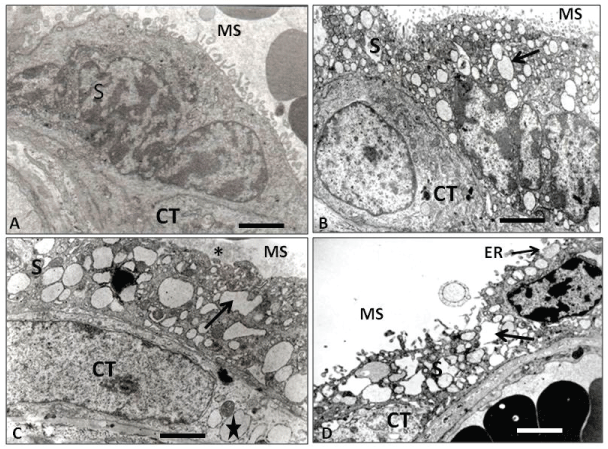
Figure 3: Electron microscopy of placental barrier. (A) Control placental villi showing the syncytium (S) toward the maternal space (MS), lying on a citotrophoblast (CT) of normal ultrastructural aspect. Dilated cisternae of RE are not seen in the syncytium; (B) IURG placental villi showing the syncytium (S) toward the maternal space (MS) with normal microvilli and intracytoplasmic dilated ER (arrow). Note that inside this epithelial layer it is possible to observe a cytotrophoblast (CT) with a normal structural appearance; (C) PE placental villi showing the syncytium (S) toward the maternal space (MS) with irregular and dilated cisternae of the ER (arrow) and loss of the upper microvilli (asterisk). Note a single cytotrophoblast (CT) underlying the syncytiotrophoblast (ST) with many ditated cisternae ER (star); (D) PE+IURG placental villi showing an apoptotic syncytium (S) with irregular and destroyed dilated ER cisternae, release of microparticules into the maternal space, ER exposed to the maternal space (ER), and the presence of an apoptotic cytotrophoblast (CT). Calibration bars 2.4 μm.
View Figure 3
IURG placentas
All IUGR cases showed a positive reaction to the AA antibody at the basal lamina level of the trophoblast stem and exchange villi, although less marked in the latter (Figure 1B). These amyloid deposits revealed the tortuous arrangement of the basal lamina between the epithelium and the underlying stroma. A positive reaction to the AA antibody was also observed in areas with stromal fibrinoid deposits, as was also noticed in the control (data not shown). No amyloid intracytoplasmic deposits were evidenced in the ST.
TEM level analysis of IUGR placentas evidenced that the exchange villi ST showed dilated ER cisternae and an underlying CT displaying normal cytological features (Figure 3B).
PE placentas
The immunohistochemistry analysis showed the presence of amyloid deposits in the stroma that, in a similar way to the IUGR cases, was also mixed with fibrinoid deposits in the stem chorionic villi. It should be emphasized that in PE placentas both the ST and CT of the chorionic exchange and the stem villi showed intracytoplasmatic amyloid deposits (Figure 1C). In addition, in almost all the syncytial nodes presented in the hematic chamber, unusual AA deposits were evidenced (Figure 1C). PE placentas treated with the anti-active Caspase 3 antibody showed positive reaction both in the CT and the ST (Figure 2B).
The TEM study showed that the ER displayed dilated cisternae in the ST, as usual. Surprisingly, such dilated cisterns were also observed in the CT, in concomitance with a significant decrease in the ST apical microvilli (Figure 3C).
PE+IUGR placentas
The immunohistochemistry study confirmed that PE+IUGR placentas showed the presence of amyloid deposits in the interior of the stem villi (endothelium) and in the ST of the stem and exchange villi. Furthermore, marked amyloid deposits in the cytoplasm of the ST stem villi were also observed. Such deposits were considerably less abundant in the exchange villi, especially in areas where extremely thin ST was evidenced, accompanied by abundant detachment of irregular syncytial debris in the hematic chamber. In these samples the amyloid deposits were also observed at the level of the fetal endothelium.
The TEM analysis revealed exchange villi in advanced stages of apoptosis, with detachments of the apical region of the ST towards the hematic chamber, the latter probably in conjunction with amyloid deposits accumulated in dilated and irregular ER cisternae, indicating the detachment of large quantities of ST microparticles and aponecrotic or inmature knots, in agreement with previous reports [38,39]. In addition, an underlying CT with a complete apoptotic aspect (Figure 3D) was also observed.
Discussion
The results reported here indicate that using immunohistochemistry and the primary anti-amyloid A antibody (Figure 1B, Figure 1C and Figure 1D) we have identified for the first time the presence of these amyloid deposits in specific areas of stem and exchange villi in placentas with either IUGR, PE or PE+IUGR. It is noteworthy that in the cases of IUGR, the amount of amyloid deposits in the exchange villi decreases as the branching of this type of villi are located far away from the stem villi. We therefore postulate that the amount of amyloid deposits varies accordingly with the specific type of pathology. From our point of view, these deposits are related to the placental ER stress, which is supported by the evidenced obtained at TEM level (Figure 3B, Figure 3C and Figure 3D). Our results are consistent with the study of Yung, et al. [31], who also observed ST cistern dilation in IUGR and PE+IUGR placentas at TEM level.
The presence of AA amyloid in placental villi substantiates the classification of the PE condition as a secondary reactive systemic amyloidosis of the inflammatory type, where the serum protein SAA is responsible for the formation of the first fibrillar cores, which ultimately will constitute the amyloid deposits, a process initially induced by the effect of cytokines [40]. This secondary amyloidosis differs from the primary form by the type of fibrillar protein deposited; in the primary amyloidosis this corresponds to the light chain of the fibrillar protein immunoglobulin, and therefore is classified as a gammopathy. It should be mentioned that there is also a third type of amyloidosis, called prealbumin, a form of hereditary polyneuropathy also defined as transthyretin amyloid (ATTR), which produces cardiac pathologies that are often confused with coronary heart disease [40,41]. ATTR has also been identified in IUGR, PE and PE+IUGR placentas [39], so it is reasonable to infer that placental amyloid deposits are composed of different types of misfolded proteins. Also noteworthy are the studies of Kalkunte, et al. [12] and Cheng, et al. [14], who using TTR antibody in paraffin sections, showed the presence of amyloid aggregates in PE placentas as well as in the serum of these patients. In the latter study, amyloid accumulation was observed in the cytoplams of the ST and intravellositarily, mixed with fibrinoid deposits, as was the case in the present study.
It is important to note that our immunohistochemical results in PE+IUGR placentas show the same arrangement of amyloids TTR deposits in the ST of the villus exchange (Figure 1D), as it was observed by Fruscalzo, et al. [42] in PE+IUGR cases, where these deposits were located in the syncytium cytoplasm, with higher concentration towards the apical edge of the syncytium, which makes the amyloid more likely to be expelled toward the hematic chamber, depending on the degree of apoptosis experienced by the ST. It is also important to mention that in our IUGR cases (Figure 1B), the AA deposits were observed in the basal zone of the ST as extracellular matrix (ECM) deposited on the trophoblast basal lamina: ie, in this epithelial layer the amyloid is expelled from its intracytoplasmic location to the basal area, where the trophoblast basal lamina is located. This fact differs from that of Fruscalzo, et al. [42] who in IUGR found the TTR amyloid to be located toward the apical syncytiotrophoblast.
The accumulation of misfolded proteins in the ER is defined as ER stress which, at TEM level, is evidenced as dilated cisternae of this organelle [26]. In this scenario the UPR cellular response is activated [23,25,27,30] and one of two possibilities will occur: i) the ER will beging secreting well folded proteins again, or ii) the cell will enter into apoptosis [20,28,29,32]. The activity of the UPR in placenta has been specifically studied by Burton, et al. [23] who found an increase in the UPR activity in IUGR placentas which was even more evident in cases of IUGR+PE versus control placentas.
In agreement with the above, in our TEM study the control placentas showed exchange villi where the placental barrier evidenced no dilated ER cisternae in the ST (Figure 3A), concomitant with the presence of underlying CT displaying normal cytological features. In our IUGR placentas, however, the ST evidenced several ER dilated cisternae, while the CT showed a normal appearance (Figure 3B). The apoptosis in the normal CT is initiated by its fusion into the ST by activation of caspasa 8, therefore an inactive form of procaspase 3 would concomitantly be integrated into the ST, which will finally leads to the ST apoptosis. This has been supported by the studies of Black, et al. [43] and Huppertz, et al. [44-47].
As it has been proposed, by some way yet to be determined [23,25,27], the presence of misfolded proteins in the ST would activate and prompt the UPR in the ST to normalize protein synthesis. Under these conditions, normal ST apoptosis would be reactivated by activation of Caspase 3 with expulsion of normal syncytial knots towards the hematic chamber. Thus, in IUGR placentas, the ST would be able to expel the misfolded proteins to the trophoblast basal region, placing them in the basal lamina as ECM secretion, preventing the excretion of the misfolded proteins to the hematic chamber where the maternal blood circulates. This extracellular accumulation of misfolded proteins in the basal lamina of the trophoblast stem villi, somehow hinders the normal mother/fetus metabolic exchange which, in part, would explain the birth of a IUGR baby. However, if the CT evidences dilated ER by deposits of misfolded proteins, as observed at TEM level in PE placentas in our study (Figure 3C), this could be interpreted as apoptosis of the CT will start previously to the fusion with the ST by action of the activation of caspase 3 in the CT (Figure 2B). Under these conditions, as a consequence of its fusion with ST, an increase of misfolded proteins and greater activity of caspase 3 will occur in the ST. As a consequence, the UPR will divert its activity to apoptosis of the ST [28,29] expelling immature or aponecrotic syncytial knots [39] towards the hematic chamber. The increase of apoptosis in the ST would produce expulsion of misfolded proteins to the hematic camera, either as ST debris or as syncytial aponecrotic nodes (Figure 1C) [39], structures that would produce PE syndrome in the mother [23]. The evidence showed in our figure 3C reinforces this notion, as it shows that the apical part of the syncytium no longer exists, which may mean that it has been expelled towards the hematic chamber. These results are consistent with the study of Goswami, et al. [33], who demonstrated the presence of large amounts of ST microparticles in the hematic chamber in PE placentas, but not in the case of IUGR.
In the present work we have found that, in PE+IUGR placentas, some villi showed an extremely thin ST without presence of amyloid deposits. We also observed syncytial remains in the hematic chamber that showed amyloid inside (Figure 1D). The absence of amyloid in the ST would suggest that the intracytoplasmic deposits have already been expelled either to the hematic chamber and/or into the stroma of the chorionic villi, in the latter case reaching the endothelium of the fetal blood vessels (Figure 1D). The consequence of amyloid deposit in the endothelium of a stem villi small artery will be the alteration of the normal functioning of the blood vessel and its branching to capillaries in the exchange villi, which consequently will affect the function of the placental triade; smooth muscle cells-telocytes-myofibroblast [48,49], thus affecting the mother/fetus metabolic exchange. In addition to that, at TEM level we observed that in PE+IUGR placentas, the ER stress has produced apoptosis in both the ST and the CT (Figure 3D). This fact further supports our hypotheses: an apoptotic CT possibly favors UPR activation to apoptosis in the ST and this would explain the presence of amiloyd, detected by proteomic screen, in the serum of women with PE, as it has been described by Kalkunte, et al. [12]. From our point of view, the amyloid deposits would depend on the amount and preservation of the parameters of normality evidenced by CT cells in the placental barrier [46]. Additionally, Veerbeek, et al. [50] demonstrated that placental ER stress was induced during the labor process, however, it is interesting to note that in our study all PE cases correspond to Caesarean-sections (Table 1), hence, these amyloid deposits are consequence of ER stress produced in the syncytium during gestation. On the other hand, McCarthy, et al. [51] and Buhimschi, et al. [13] using Congo red, a dye with high affinity for amyloidophilic structures, have determined the presence of different types of amyloid in the urine of women with PE and IUGR. Both groups of researchers have proposed that the congophilia detected in the urine of these patients could be a good predictor index of PE. From our point of view, in both cases this situation would depend on the amount and preservation of the parameters of normality evidenced by CT cells in the placental barrier [52]. It is also important to note that Kalkunte, et al. [53] previously demonstrated that administration of serum from women with PE to mice IL-10 -/-, reproduced the full spectrum of preeclampsia-like symptoms, caused hypoxic injury in uteroplacental tissues, and elevated soluble fms-like tyrosine kinase 1 and soluble endoglin, markers thought to be related to the disease. The same authors showed latter that native TTR, ie TTR with normal folding, inhibits all preeclampsia-like features in the humanized mouse model [12]. It is interesting to add that our PE cases came all from Caesarea-sections and therefore the AA deposit is consequence of ER stress produced in the syncytium, therefore, is not comparable to the study by Veerbeek, et al. [50] who demonstrated that the increase in ER stress was induced during the labor process.
The results of our TEM study are also consistent with those of Yung, et al. [31] and Burton and Yung [54]. Our contribution aims to reinforce the studies of these authors adding now the hypothesis that the degree of apoptosis experienced by the CT prior to its fusion into the ST could be considered to link the degree of ER stress experienced by ST and with the destination of the infoldings proteins produced in the process.
Conclusions
We postulate here that the CT regulates, probably through the UPR in the ST, the accumulation of misfolded proteins and their expulsion either towards the hematic chamber or to the trophoblast basal lamina. The excretion of misfolded proteins towards the hematic chamber, either as ST apoptotic microparticles or syncytial aponecrotic nodes, would cause the patient to develop PE symptoms. On the contrary, if these proteins accumulate in the basal lamina of the trophoblast, the impairment of the mother/fetus metabolic exchange due to an alteration of the functional triade would explain in part the birth of an IUGR baby. Finally, if both situations occur, ie excretion of misfolded proteins into the hematic chamber followed by its deposit beyond the basal lamina, as in the fetal endothelium, PE+IUGR will be developed.
The morphological evidence provided in our work regarding the deposit of amyloid in the ST and its subsequent expelling into the hematic chamber as ST microparticles allows us to support the assertions of Kalkunte, et al. [12], Buhimschi, et al. [13] and Chen, et al. [14], regarding that PE could be considered in the group of the so called "conformational diseases".
Projections
The determination of ST microparticles using flow cytometry in the serum or urine of pregnant women, together with the use of fluorescent immunohistochimestry will provide valuable information about the degree of severity in cases of PE, an idea also proposed by McCarthy, et al. [51], Buhimschi, et al. [13] and Jonas, et al. [55], but only verified at molecular level.
Conflict of Interest
None.
Acknowledgmets
This research was partly supported by Fondo Nacional de Desarrollo Científico y Tecnológico (FONDECYT), grants 1050482 and 1090245, and was approved by the Bioethic Committee of the Faculty of Medicine and the Clinical Hospital. All women were required to sign an informed consent to participate in this study. We gratefully acknowledge the technical assistance of Ricardo Contreras and the technical ultrastructural assistance of CESAT. Finally, the authors wish to thank Miss Paulina González Díaz for her kind assistance with language editing.
References
-
Roberts JM, Cooper DW (2001) Pathogenesis and genetics of pre-eclampsia. Lancet 357: 53-56.
-
Redman CW (1991) Current topic: pre-eclampsia and the placenta. Placenta 12: 301-308.
-
Bosco C, González J, Gutiérrez R, Parra-Cordero M, Barja P, et al. (2012) Oxidative damage to pre-eclamptic placenta: immunohistochemical expression of VEGF, nitrotyrosine residues and von Willebrand factor. J Matern Fetal Neonatal Med 25: 2339-2345.
-
(1990) National High Blood Pressure Education Program Working Group Report on High Blood Pressure in Pregnancy. Am J Obstet Gynecol 163: 1691-1712.
-
Davey DA, MacGillivray I (1988) The classification and definition of the hypertensive disorders of pregnancy. Am J Obstet Gynecol 158: 892-898.
-
Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, van Asshe A (1994) A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol 101: 669-674.
-
Roberts JM, Hubel CA (1999) Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet 354: 788-789.
-
Khong TY, De Wolf F, Robertson WB, Brosens I (1986) Inadequate maternal vascular response to placentation in pregnancies complicated by pre-eclampsia and by small-for-gestational age infants. Br J Obstet Gynaecol 93: 1049-1059.
-
Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ (2002) Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90: 1274-1281.
-
Sibai BM (2007) Biomarker for hypertension-preeclampsia: are we close yet? Am J Obstet Gynecol 197: 1-2.
-
Redman CW, Sargent IL (2009) Placental stress and pre-eclampsia: a revised view. Placenta 30: S38-42.
-
Kalkunte S, Neubeck S, Norris WE, Cheng SB, Kostadinov S, et al. (2013) Transthyretin is dysregulated in preeclampsia, and its native form prevents the onset of disease in a preclinical mouse model. Am J Pathol 183: 1425-1436.
-
Buhimschi IA, Nayeri UA, Zhao G, Shook LL, Pensalfini A, et al. (2014) Protein misfolding, congophilia, oligomerization, and defective amyloid processing in preeclampsia. Sci Transl Med 16: 245-292.
-
Cheng SB, Nakashima A, Sharma S (2016) Understanding Pre-Eclampsia Using Alzheimer's Etiology: An Intriguing Viewpoint. Am J Reprod Immunol 75: 372-381.
-
Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4: 49-60.
-
Soto C, Estrada LD (2008) Protein misfolding and neurodegeneration. Arch Neurol 65: 184-189.
-
Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384-396.
-
Moreno-Gonzalez I, Soto C (2011) Misfolded protein aggregates: Mechanisms, structures and potential for disease Transmission. Semin Cell Dev Biol 22: 482-487.
-
Yoshida H (2007) ER stress and diseases. FEBS J 274: 630-658.
-
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115: 2656-2664.
-
Malhotra JD, Kaufman RJ (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18: 716-731.
-
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9: 2277-2293.
-
Burton GT, Yung HW, Cindrova-Davies T, Charnock-Jones DS (2009) Placental Endoplasmic Reticulum Stress and Oxidative Stress in the Pathophysiology of Unexplained IntrauterineGrowth Restriction and Early Onset Preeclampsia. Placenta 30: 43-48.
-
Yung HW, Korolchuk S, Tolkovsky AM, Charnock-Jones DS, Burton GJ (2007) Endoplasmic reticulum stress exacerbates ischemia- reperfusion-induced apoptosis through attenuation of Akt protein synthesis in human choriocarcinoma cells. FASEB J 21: 872-884.
-
Cao SS, Kaufman RJ (2014) Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal 21: 396-413.
-
Schröder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74: 739-789.
-
Szegezdi E, Logue SE, Gorman AM, Samali A (2006) Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 7: 880-885.
-
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7: 1013-1030.
-
Fribley A, Zhang K, Kaufman RJ (2009) Regulation of apoptosis by the unfolded protein response. Methods Mol Biol 559: 191-204.
-
Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C (2013) When ER stress reaches a dead end. Biochim Biophys Acta 1833: 3507-3517.
-
Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, et al. (2008) Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. Am J Pathol 173: 451-462.
-
Rasheva VI, Domingos PM (2009) Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 14: 996-1007.
-
Goswami D, Tannetta DS, Magee LA, Fuchisawa A, Redman CW, et al. (2006) Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta 27: 56-61.
-
Bosco C, Parra M, Barja P, Rodrigo R, Fernández V, et al. (2005) Increased immunohistochemical expression of thrombomodulin at placental perivascular myofibroblast in severe preeclampsia (PE). Histol Histopathol 4: 1045-1055.
-
Parra M, Rodrigo R, Barja P, Bosco C, Fernández V, et al. (2005) Screening test for preeclampsia through assessment of uteroplacental blood flow and biochemical markers of oxidative stress and endothelial dysfunction. Am J Obstet Gynecol 193: 1486-1491.
-
Juez G (1989) Intrauterine growth curve for the appropriate diagnosis of intrauterine growth retardation. Rev Med Chil 117: 1311.
-
Bosco C (1994) Morphology of the capillaries in the alpha and beta zone of human term placenta: the relationship between capillary morphology and the trophoblastic layer. Med Sci Res 22: 115-117.
-
Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, et al. (2000) Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol 182: 41-49.
-
Huppertz B, Kingdom J, Caniggia I, Desoye G, Black S, et al. (2003) Hypoxia favours necrotic versus apoptotic shedding of placental syncytiotrophoblast into the maternal circulation. Placenta 24: 181-190.
-
Pepys MB (2006) Amyloidosis. Annu Rev Med 57: 223-241.
-
Westermark P, Wernstedt C, Wilander E, Hayden DW, O'Brien TD, et al. (1987) Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci USA 84: 3881-3885.
-
Fruscalzo A, Schmitz R, Klockenbusch W, Köhler G, Londero AP, et al. (2012) Human placental transthyretin in fetal growth restriction in combination with preeclampsia and the HELLP syndrome. Histochem Cell Biol 138: 925-932.
-
Black S, Kadyrov M, Kaufmann P, Ugele B, Emans N, et al. (2004) Syncytial fusion of human trophoblast depends on caspase 8. Cell Death Differ 11: 90-98.
-
Huppertz B, Kadyrov M, Kingdom JC (2006) Apoptosis and its role in the trophoblast. Am J Obstet Gynecol 195: 29-39.
-
Huppertz B, Frank HG, Kingdom JC, Reister F, Kaufmann P (1998) Villous cytotrophoblast regulation of the syncytial apoptotic cascade in the human placenta. Histochem Cell Biol 110: 495-508.
-
Gauster M, Huppertz B (2010) The paradox of caspase 8 in human villous trophoblast fusion. Placenta 31: 82-88.
-
Huppertz B, Frank HG, Reister F, Kingdom J, Korr H, et al. (1999) Apoptosis cascade progresses during turnover of human trophoblast: analysis of villous cytotrophoblast and syncytial fragments in vitro. Lab Invest 79: 1687-1702.
-
Bosco CB, Díaz EG, Gutierrez RR, González JM, Parra-Cordero M, et al. (2016) Placental Hypoxia Developed During Preeclampsia Induces Telocytes Apoptosis in Chorionic Villi Affecting The Maternal-Fetus Metabolic Exchange. Curr Stem Cell Res Ther 11:420-425.
-
Bosco C, Díaz E (2016) Presence of Telocytes in a Non-innervated Organ: The Placenta. Adv Exp Med Biol 913: 149-161.
-
Veerbeek JH, Tissot Van Patot MC, Burton GJ, Yung HW (2015) Endoplasmic reticulum stress is induced in the human placenta during labour. Placenta 36: 88-92.
-
McCarthy FP, Adetoba A, Gill C, Bramham K, Bertolaccini M, et al. (2016) Urinary congophilia in women with hypertensive disorders of pregnancy and preexisting proteinuria or hypertension. Am J Obstet Gynecol 215: 464-464.
-
Longtine MS, Chen B, Odibo AO, Zhong Y, Nelson DM (2012) Villous trophoblast apoptosis is elevated and restricted to cytotrophoblasts in pregnancies complicated by preeclampsia, IUGR, or preeclampsia with IUGR. Placenta 33: 352-359.
-
Kalkunte S, Boij R, Norris W, Friedman J, Lai Z, et al. (2010) Sera from preeclampsia patients elicit symptoms of human disease in mice and provide a basis for an in vitro predictive assay. Am J Pathol 177: 2387-2398.
-
Burton GJ, Yung HW (2011) Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertens 1: 72-78.
-
Jonas SM, Deserno TM, Buhimschi CS, Makin J, Choma MA, et al. (2016) Smartphone-based diagnostic for preeclampsia: a mHealth solution for administering the Congo Red Dot (CRD) test in settings with limited resources. J Am Med Inform Assoc 23: 166-173.
