Synovitis is common in knee OA patients and a known contributor to disease incidence and progression. Macrophages are the most common immune cell type present in this inflamed synovial tissue and expectedly contribute both directly and indirectly to OA progression through the induction of inflammatory mediators, growth factors and proteinases, resulting in enhanced cartilage degeneration and osteophyte formation. Furthermore, macrophage infiltration and soluble macrophage products may be associated with pain in OA, although data are somewhat controversial. OA synovium features macrophage heterogeneity, represented by the presence of subsets covering the broad spectrum of M1 (pro-inflammatory) to M2 (anti-inflammatory) phenotypes, whose emergence corresponds to the cytokine profile found in OA patients. Conflicting results have been reported in both early and advanced OA concerning macrophage number, location, marker expression and cytokine profile, which may also be a consequence of the lack of universal definitions of disease stages and OA phenotypes. In this review, we summarize and discuss the positioning of synovial macrophages in knee OA in the context of pathogenesis, synovitis, and disease monitoring and as a target for therapeutic interventions.
Macrophage, Osteoarthritis, Knee, Synovitis
ACLT: Anterior Cruciate Ligament Transaction; BMP: Bone Morphogenic Protein; CCL: CC-chemokine Ligand; CXCL: CXC-Chemokine Ligand; COX: Cyclooxygenase; DAMP: Damage-Associated Molecular Pattern; DMM: Destabilized Medial Meniscus; DMOAD: Disease-Modifying Osteoarthritic Drug; ECM: Extracellular Matrix; FPR: Formyl Peptide Receptor; FR: Folate Receptor; GM-CSF: Granulocyte Macrophage Colony-Stimulating Factor; IFN-γ: Interferon γ; IL: Interleukin; iNOS: Inducible Nitric Oxide Synthase; KL grade: Kellgren-Lawrence Radiographic Grade of OA; LPS: Lipopolysaccharide; M-CSF: Macrophage Stimulating Factor; MIA: Monosodium Iodoacetate; MIF: Macrophage Migration Inhibitory Factor; MMP: Matrix Metalloproteinase; MRI: Magnetic Resonance Imaging; MTX: Methotrexate; NF-κβ: Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B cells; NGF: Nerve Growth Factor; NO: Nitric Oxide; NSAIDs: Nonsteroidal Anti-Inflammatory Drugs; OA: Osteoarthritis; PET: Positron Emission Tomography; RA: Rheumatoid Arthritis; RANKL: Receptor Activator of NF-κB ligand; ROS: Reactive Oxygen Species; SPECT: Single-Photon Emission Computed Tomography; TA: Triamcinolone Acetonide; Th: T Helper Cell; TGF: Tumor Growth Factor; TLR: Toll-Like Receptor; TNF: Tumor Necrosis Factor; US: Ultrasonography
Osteoarthritis (OA) is among the ten leading causes of disability in the Western world [1-3], affecting the elderly as well as individuals of working age [4]. OA is generally a slowly progressive joint disease and is characterized by pain, stiffness, and sometimes swelling of the joint [5,6], all of which can result in impaired function [1,3]. Current non-surgical treatment options focus on symptom alleviation, including analgesics, anti-inflammatory therapy and physical and occupational therapy. However, these treatments do not suffice as they do not modify the course of the underlying disease [7,8]. Hence, due to the high economic and personal burden related to OA, there is a large and growing demand for disease-modifying therapies (DMOADs).
To date, the pathogenesis of OA has not been elucidated completely. Previously, OA was merely considered a disease of ‘wear and tear' [9-12] with cartilage degradation as its key characteristic [3]. Nowadays, OA is considered a multifactorial disease affecting the whole organ (Figure 1), resulting in structural and functional changes in many articular and periarticular tissues [2,9,10,13]. Moreover, OA is considered a heterogeneous disease, leading to a variety of disease manifestations between patients, joints, and disease stages [2,9,10,13,14]. Due to this heterogeneity, it is hypothesized that stratification of OA subtypes is warranted in order to provide appropriate treatments.
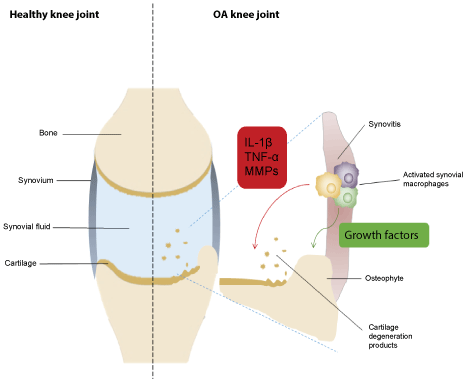 Figure 1: Schematic overview of a healthy knee joint (left) and osteoarthritic knee joint (right). The cardinal features of OA are cartilage damage, osteophyte formation and subchondral sclerosis, and synovitis. Synovial macrophages play a critical role in cartilage degeneration and osteophyte formation by the induction of pro-inflammatory cytokines (e.g. TNF-α and IL-1β), growth factors and matrix metalloproteinases (MMPs), resulting in clinical symptoms of OA. Abbreviations: IL-1β, interleukin-1β; TNF-α, tumor necrosis factor α. View Figure 1
Figure 1: Schematic overview of a healthy knee joint (left) and osteoarthritic knee joint (right). The cardinal features of OA are cartilage damage, osteophyte formation and subchondral sclerosis, and synovitis. Synovial macrophages play a critical role in cartilage degeneration and osteophyte formation by the induction of pro-inflammatory cytokines (e.g. TNF-α and IL-1β), growth factors and matrix metalloproteinases (MMPs), resulting in clinical symptoms of OA. Abbreviations: IL-1β, interleukin-1β; TNF-α, tumor necrosis factor α. View Figure 1
Synovitis is a common feature in OA, but its severity and timing differ among patients [2,5,10,12,13,15-18]. Synovitis does seem to correspond to pain [10]. Furthermore, several studies have demonstrated an association between synovitis and (radiographic) OA progression [2,5,9,16] in general and the advancement of cartilage breakdown in particular [12,18,19]. Therefore, treating synovitis in OA would potentially be beneficial, as both symptom alleviation and reduction of joint degradation could be achieved [12]. To this end, DMOADs may serve as effective treatments of synovitis [12,16].
A possible treatment strategy for OA could be the modulation of infiltrating cells. In rheumatic joints, neutrophils and lymphocytes generally comprise the majority of leukocytes and this is reflected in the synovial fluid in active rheumatoid arthritis (RA) [20]. In contrast, macrophages are relatively more present in the synovial fluid in OA as compared to RA [21]. Furthermore, macrophages are also found to be the predominant inflammatory cell type in the synovial tissue in OA [12,22,23]. Their role in OA, however, is less well understood.
Macrophages represent a dynamic cell type harboring the ability to alter their pro- or anti-inflammatory phenotype and function upon environmental stimuli [24]. Several studies have demonstrated that synovial macrophage infiltration is positively correlated with OA progression and disease severity [25,26]. Moreover, synovial macrophages have been associated with cartilage degradation [27], osteophyte formation [28], and pain [29] in OA (Figure 1). As macrophages are key cells in the pathogenesis of OA, modulating synovial macrophages might be sufficient to alleviate OA symptoms and prevent progression.
The potential role of synovial macrophages in knee OA will be further discussed in the following paragraphs of this review. To ensure a comprehensive overview a literature (Supplementary File) search strategy was employed as described in Supplementary Figure 1.
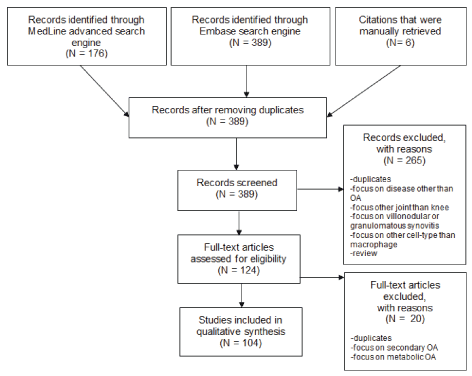 Supplementary Figure 1: Overview of the step-wise selection of records and numbers of included/excluded articles. View Supplementary Figure 1
Supplementary Figure 1: Overview of the step-wise selection of records and numbers of included/excluded articles. View Supplementary Figure 1
The synovial membrane constitutes a boundary between the joint and surrounding musculoskeletal tissues [30]. Through the secretion of synovial fluid, synovium conserves articular mobility and limits friction between articular cartilage surfaces [6,31]. Additionally, the synovial fluid nourishes the articular cartilage and removes metabolites and other matrix degradation products from the joint [12].
The synovium consists of two layers: the synovial lining or intima, comprising 2 to 3 cell layers of synovial macrophages (type A synoviocytes) and fibroblasts (type B synoviocytes); and the synovial sublining or subintima, containing collagen type III, blood vessels and a few immune cells, including macrophages [6,30-32]. Synovial fibroblasts primarily produce synovial fluid components, including hyaluron [30,31], whereas synovial macrophages remove joint debris from the synovial fluid, clear bacterial infections, and control the pro-inflammatory and anti-inflammatory cytokine balance in the synovial fluid [30]. Therefore, both cell types are vital for the maintenance of joint homeostasis.
Multiple methodologies are available for detecting and grading of synovitis in OA and other joint diseases, including arthroscopy, synovial biopsy, and non-invasive imaging techniques such as magnetic resonance imaging (MRI) and ultrasonography (US) [16]. Synovitis is detected in 50% of the patients using arthroscopy [18] and in 47% and 73% of the patients using US and MRI, respectively [12]. Typically, synovitis cannot be visualized by X-ray imaging.
Arthroscopy portrays synovitis as localized proliferative alterations in the synovium with signs of inflammation, such as an increased vascularity [12]. MRI can be used to determine the presence and/or extent of synovial thickening and effusion [12,33]. US can be used to monitor synovial hypertrophy, hyperemia, and joint effusion [12,19]. The latter non-invasive techniques are increasingly used for assessing synovitis, as their findings correlate to arthroscopic findings [12,16] and/or histopathologically observed synovitis [12,33].
Histological aberrancies in the OA synovium include synovial hypertrophy and hyperplasia, accompanied by infiltration of mononuclear cells in the synovial sublining and lining layer [11,12,34]. Inflamed areas in the OA synovium show increased vascularization and cellular infiltration [18]. Generally, synovitis is found in close proximity to damaged cartilage [12,18,35], but no preferential location has been found so far [16].
The extent of synovitis differs between disease severity levels, disease stages, and patients [16]. In general, synovitis is present in both early and advanced OA, but tends to increase with disease severity [16,19,36,37]. Studies by Ayral, et al. demonstrated that in advanced OA, characterized by excessive cartilage damage, synovitis detected by arthroscopy was more prominent as compared to early-stage OA [18]. However, other studies have failed to detect synovitis [36,37] or showed synovitis in less than 50% of advanced OA patients [38]. Moreover, synovitis can precede the radiographic presentation of knee OA [39,40], indicating that synovitis might even predate cartilage change [39]. These findings underline the great variety in synovitis between OA patients, but at the same time its potential importance.
Synovial macrophages are located in the synovial lining and scattered throughout the synovial sublining in OA, mostly confined to sites of cartilage damage [26,37,41-43]. Even though synovial macrophage numbers are lower in OA than in RA [44], macrophages are the most prominent immune cell type [34,45-47] and are highly activated [29,42] in OA.
Animal studies by Blom, et al. suggest that macrophage activation can occur due to cartilage damage in OA [28,48]. Mediators from cartilage probably leak into the synovial fluid and activate synovial macrophages (Figure 1). Potential mediators include damage-associated molecular patterns (DAMPs), including extracellular matrix (ECM) components released by damaged cartilage, e.g. fibrinogen and plasma proteins, such as α1m, α2m and Gc-globulin [49], alarmins [50], and basic calcium phosphate crystals [49,51]. DAMPs predominantly signal via Toll-like receptor (TLR) 4, expressed by synovial macrophages, and CD14, which forms a complex with TLR-2 and TLR-4 to initiate DAMP-related macrophage activation [25,52]. In OA, the expression of macrophage markers, including CD14 and MHC class II genes, is associated with joint space narrowing and osteophyte formation [26].
Activation of synovial macrophages leads to release of tumour necrosis factor (TNF)-α and other pro-inflammatory cytokines [49,51], catabolic mediators, such as matrix metalloproteinases (MMPs) [12,28], and anabolic factors, which can induce osteophyte formation [28]. The notion that release of cartilage breakdown products can trigger activation of macrophages and cytokine production indicates that synovitis is part of a vicious cycle of inflammation and cartilage breakdown [53]. In this cascade of events, the activation mechanism of macrophages in OA and their role as either initiator and/or drivers of OA remains to be defined.
Activated synovial macrophages in OA can also stimulate other immune cells, particularly T cells. In fact, the mononuclear infiltration is mainly comprised of CD4+ T cells [34,45,46]. Macrophages can take up fragments of type II collagen in OA, present them on their surface, and activate T cells [35]. Studies by Shen, et al. show that CD4+ T cell numbers increase at OA onset in the anterior cruciate ligament transection (ACLT) model. Furthermore, the increased numbers of CD4+ T cells were followed by a rise in numbers of synovial macrophages, which can possibly be explained by the stimulation of CCL9 release by CD4+ T cells [54]. Together, these findings suggest a close interplay between macrophages and CD4+ T cells in OA.
Currently, synovial macrophages can only be visualized histologically. However, non-invasive tools for visualizing macrophages are under development. Macrophage Positron Emission Tomography (PET) imaging using the macrophage G-protein coupled receptor formyl peptide receptor (FPR)-1 tracer showed encouraging results in experimental OA [55]. Another approach utilizes Etarfolatide, which has a high binding affinity for the folate receptor (FR)-β expressed on activated macrophages [25]. Single-photon emission computed tomography (SPECT) with (99m)Tc-EC20 (Etarfolatide) has been successfully applied to detect macrophages in knee joints of patients with varying degrees of OA [29]. Lastly, a recent study demonstrated macrophage visualization in OA using SPECT with 111In-Octreoscan, which has a high affinity for the somatostatin receptor subtype 2 expressed by macrophages [56].
Macrophages are dynamic cells that can respond to stimuli in their microenvironment through modification of their phenotype and function [57]. Because of this, macrophages are key players in initiation as well as resolution of inflammation [57]. Although a spectrum of macrophage subsets and even mixed subtypes of macrophages exists [58], a crude subdivision can be made in two extremes: "M1" classically activated and "M2" alternatively activated macrophages (Figure 2). M2 macrophages can be further classified in subgroups, including M2a, M2b and M2c, based on activation stimuli and functions [59-61].
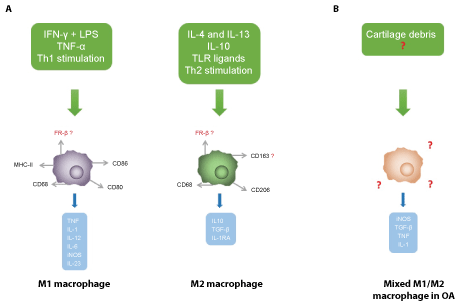 Figure 2: (A) Macrophages can be subdivided in M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophages, based on in vitro activation mechanism and immunophenotypical markers. M1 macrophages are activated by IFN-γ and LPS and by TNF-α, while M2 macrophages are activated by interleukin (IL)-4, IL-13, IL-10 and other specific TLR ligands, such as complement components. Activated M1 macrophages predominantly secrete inflammatory cytokines and catabolic mediators, whereas activated M2 macrophages release anti-inflammatory cytokines and growth factors. Nowadays, cartilage degeneration products are considered potential activators of synovial macrophages in OA, which results in (B); a mixed M1/M2 phenotype expressing distinct markers and releasing a variety of mediators.
Figure 2: (A) Macrophages can be subdivided in M1 (pro-inflammatory) and M2 (anti-inflammatory) macrophages, based on in vitro activation mechanism and immunophenotypical markers. M1 macrophages are activated by IFN-γ and LPS and by TNF-α, while M2 macrophages are activated by interleukin (IL)-4, IL-13, IL-10 and other specific TLR ligands, such as complement components. Activated M1 macrophages predominantly secrete inflammatory cytokines and catabolic mediators, whereas activated M2 macrophages release anti-inflammatory cytokines and growth factors. Nowadays, cartilage degeneration products are considered potential activators of synovial macrophages in OA, which results in (B); a mixed M1/M2 phenotype expressing distinct markers and releasing a variety of mediators.
Abbreviations: FR β: Folate Receptor β; IFN-γ: Interferon γ; IL: Interleukin; IL-1RA: IL-1 Receptor Antagonist; iNOS: inducible Nitric Oxide Synthase; LPS: Lipopolysaccharide; TGF-β: Tumor Growth Factor β; Th1/2: T Helper Cell Type 1/2; TNF: Tumor Necrosis Factor; TLR: Toll-like Receptor. View Figure 2
M1 and M2 macrophages exert different functions, related to their distinct cytokine release profile. M1 macrophages are involved in Th1 stimulation and are characterized by IL-12, IL-23, TNF-α, IL-1 and nitric oxide (NO) release. M2 macrophages stimulate Th2 cells [42,60] and predominantly release IL-10, CCL18, TGF-β and IL-1 receptor antagonist (IL-1RA) [42,57,61] upon activation (Figure 2). In turn, M1 and M2 macrophages are activated through cytokines produced by Th1 and Th2 cells, respectively [42]. M1 activation occurs through interferon γ (IFN-γ), the TLR ligand lipopolysaccharide (LPS) and components of intracellular bacteria. Skewing to M2 macrophages proceeds upon exposure to interleukin IL-4, IL-13, parasites, complement factors, macrophage stimulating factor (M-CSF), IL-10 and tumor growth factor (TGF)-β [42,57,59] (Figure 2).
Originally, M1 macrophages were considered pro-inflammatory, while M2 macrophages were considered anti-inflammatory through production of anti-inflammatory and regulatory cytokines and chemokines [60]. This gave rise to the assumption that M2 macrophages are beneficial in inflammatory diseases and might be involved in tissue repair and wound healing [59]. However, this view has been (partially) revisited as M2 macrophages have also been associated with chronic inflammation in cancer and other diseases [59,62] and have been shown to be able to produce pro-inflammatory cytokines in a synovial microenvironment with auto-antibodies in RA patients [63,64].
In addition to their distinct function and cytokine release, M1 and M2 macrophages can be distinguished through differentially expressed receptors and other markers. CD14 [26], a marker of monocytes and macrophages, is more abundantly expressed on M2 compared to M1 macrophages [25]. CD163 [25,57,62,65,66] and CD206 [67] are considered preferred markers of M2 macrophages. M1 macrophages predominantly express MHC class II, CD68, CD80 and CD86 [66,68] (Figure 2).
Although Folate Receptor β (FRβ) expression has been identified as a marker for activated macrophages in RA [69-71], its expression on polarized macrophages is incompletely known; in tumour-associated macrophages FRβ is expressed on M2 macrophages [72], but in OA it is also expressed on M1 macrophages [42]. Thus, caution should be taken when classifying FRβ+ macrophages as M2 macrophages solely on the basis of CD163 co-expression as both receptors seem to have a mixed pattern of expression on M1 and M2 macrophages in RA and OA [42].
The inflammatory milieu of the synovium in OA has been implicated in the skewing of macrophages to both pro-inflammatory and anti-inflammatory phenotypes. In this respect, Utomo, et al. demonstrated that most synovial macrophages in OA have a M2 phenotype characterized by IL-10 production [62]. However, multiple studies demonstrated that both M1 and M2 macrophage phenotypes are present in OA [24,28,41,66,68,73,74]. In fact, synovial macrophages in OA were identified to actually express a combination of M1 and M2 phenotypic markers, including CD14, CD163 and CD86 [24,34,36,73,75-77] (Figure 2). It has been hypothesized that the combined expression of both M1 and M2 macrophage markers might be caused by synovial macrophages being "trapped in transition" [25,29]. The exact mechanism underlying activation of M1 and M2 synovial macrophages in OA is still unresolved.
In early stages of OA, increased skewing of bone marrow monocyte precursors towards the M1 phenotype has been reported [74]. However, macrophages stimulated with synovial fluid of early OA resulted in increased release of CCL2 and IL6 by mesenchymal stromal cells (MSC), which in turn can direct M2 polarization of macrophages [78]. Studies by Fahy, et al. report a large intra-patient variation in phenotypic marker expression patterns between OA synovium regions. Specifically, CD86 expression was either absent or present on macrophages in the synovial lining or sublining, while expression of CD206 was most pronounced on macrophages in the lining layer [67]. Likewise, Manferdini, et al. mainly identified CD80 M1 macrophages in the OA synovium lining layer, while CD206-positive M2 macrophages were predominantly located in the sublining layer [68]. Moreover, Manferdini, et al. reported a large inter-patient variability in the M1 and M2 marker expression in the OA synovium [68]. Hence, generalizations of the phenotypic marker expression patterns should be exercised with caution.
Several studies have demonstrated differences between RA and OA synovial macrophage phenotypes. Lower expression of CD14+ macrophages was found in OA as compared to RA synovium [68], whereas TGF-β expression was higher in OA [42]. Furthermore, macrophages in OA synovium had lower FRβ expression than RA macrophages, and FRβ+/CD163+ macrophages were more dominant than FRβ+/CD163- macrophages in the lining layer of the OA synovium [42]. Together, relatively high M2 marker expression is demonstrated on OA synovial macrophages although absolute expression appears lower than on RA synovial macrophages.
In general, higher inflammatory cytokine levels are observed in RA compared with OA synovium, although both diseases show partly analogous cytokine profiles, consisting of elevated levels of IL-1β, TNF-α, IL-6 and IL-4 [44,79-82]. However, there are also potentially important differences, such as the granulocyte macrophage colony-stimulating factor (GM-CSF) expression that is more prominent in OA compared with RA [44]. In OA, anti-inflammatory cytokine levels, e.g. IL-10, are increased compared to healthy controls [73], which may be consistent with the suggested dominance of M2 over M1 macrophages in OA.
IL-1β and TNF-α constitute the key mediators released by activated pro-inflammatory macrophages and staining patterns of these mediators correspond to the distribution of synovial macrophages in OA [24]. Both IL-1β and TNF-α are primarily detected in the lining layer and to a lesser extent in the sublining layer of OA synovium [24,27,83]. Furthermore, IL-1β and TNF-α induce pro-inflammatory cytokine production, including IL-6 and IL-8, and thereby stimulate inflammation. Moreover, IL-1β suppresses type II collagen, which is a key constituent of cartilage, and subsequently aggravates cartilage damage [27]. Together, these findings suggest that synovial macrophages of mixed phenotypes account for the pro-inflammatory and anti-inflammatory cytokine production to fuel the pathogenesis of OA. However, Beekhuizen, et al. did not observe elevated levels of TNF-α and IL-1β in advanced OA [84], suggesting that these cytokines may not be essential in this phase of OA.
Macrophages are not the only cell type in the joint capable of producing these indicated cytokines and the exact cellular source of remains largely unclear [24]. Furthermore, a study by Manferdini, et al. demonstrates that cell cultures containing both synovial fibroblasts and macrophages produced more pro-inflammatory mediators, including IL-6 and IL-8, compared to cell cultures with synovial fibroblasts alone [73]. This finding illustrates that macrophages are directly involved in the induction of inflammatory cytokine release from synovial fibroblasts, but mechanisms driving this process remain to be determined.
Chemokines are small secreted factors that mediate recruitment of cells [85]. Several OA studies found increased levels of chemokines involved in the attraction of macrophages. CCL2 is one of the most important chemokines involved in macrophage attraction in chronic inflammation [73,86] and elevated levels of CCL2 promote OA development [87]. CCL13 is involved in recruitment of monocytes and T lymphocytes to joints. Gao, et al. found a positive correlation between serum and synovial fluid levels of CCL13 and radiographic severity of knee OA [88]. Additionally, CXCL12 is involved in macrophage recruitment and CXCL12 plasma and synovial fluid levels are correlated with radiographic OA disease severity, which could be explained by their role in macrophage attraction [89]. Other mediators involved in macrophage attraction include CCL22, which is elevated in the synovial fluid of OA patients [84], and CCL3, of which plasma levels are positively correlated with radiographic OA disease severity [90]. Chemokines may thus mediate the attraction of macrophages to the joint and the worsening of OA.
Studies on CD68 expression suggest a pronounced macrophage infiltration in synovium of patients with early OA compared to advanced OA [73,91]. This coincided with an increased expression of nuclear factor (NF)-κβ, inducible nitric oxide synthase (iNOS) and other pro-inflammatory mediators by macrophages in early OA [39,93]. Ene, et al. reported high levels of inflammatory cytokines, including IL-6, IL-1, TNF-α, and a greater mononuclear cell infiltrate in early compared to advanced OA [92]. Consistently, Benito, et al. reported increased TNF-α, IL-1β and VEGF expression and mononuclear cell infiltration in early compared with advanced OA [91]. Lastly, Ning, et al. confirmed an increase in macrophage-related mediators in early compared to advanced OA [93].
Oppositely, other studies observed similar CD68+ macrophages numbers in synovium of early and advanced OA [83,94]. Additionally, similar synovial fluid TNF-α levels were found in early and advanced OA, whereas IL-1β expression was increased in the advanced stage OA group [83]. The reason for this apparent discrepancy in results is not clear.
It has been hypothesized that innate immunity is more prominent in early OA, while adaptive immunity dominates in advanced OA. Firstly, larger lymphoid infiltrations were observed in the synovium in advanced OA compared to early OA [38]. Secondly, the more extensive immune cell infiltration in advanced as compared to early OA could be explained by the subsequent activation of adaptive immunity. Thirdly, this hypothesis could explain the increase of mediators such as VEGF, IL-4, IL-6, IL-8, CCL3 and CCL4 in advanced stage OA and their positive correlation to radiographic severity of OA [77,80,91,95].
Several preclinical and clinical studies have shown that synovitis is linked to cartilage damage in OA [96]. Macrophages are the predominant immune cells in synovitis [27] and CD14 deficiency is correlated to delayed cartilage degradation in destabilized medial meniscus (DMM) OA models in mice [53]. Utomo, et al. studied the impact of factors released by macrophage subsets on cartilage degradation in an in vitro cartilage explant model. Their results demonstrated that pro-inflammatory M1 products stimulate cartilage damage, whereas anti-inflammatory M2 factors failed to directly inhibit cartilage degeneration or inflammation [97]. Furthermore, a recent study has shown that M1 mediators inhibit mesenchymal stem cell chondrogenic capacity and thereby exacerbate cartilage damage [68].
Inflammatory mediators released by macrophages at least partially induce cartilage damage by both downregulating matrix anabolism by chondrocytes and inducing release of MMPs, aggrecanases and other inflammatory mediators by synovial fibroblasts [77,94,98] (Figure 1). Bondeson, et al. depleted synovial macrophages by anti-CD14 conjugated magnetic beads in synovial cell cultures derived from digested OA synovium, which resulted in reduced production of IL-1β, TNF-α and MMPs by synovial fibroblasts and in less cartilage damage [22]. Furthermore, cartilage degradation was decreased in IL-1β and TNF-α neutralisation experiments [22].
Macrophages induce cartilage damage through the release of MMPs, including MMP-1, -3 and -9 [48], and through the secretion of cytokines, which in turn lead to proteinase secretion by synovial fibroblasts [99]. Depletion of macrophages from OA synovium resulted in lower expression of MMP-2, -3, and -9 [22,48], as well as CCL2 [22]. CCL2 is a chemokine involved in macrophage attraction and normally associated with increased MMP-3 and MMP-13 production [87]. MMP-3 release is also increased by TNF-α and IL-1β stimulation [100], both key mediators released by macrophages.
In addition to MMP-induced cartilage damage, activated macrophages increase cartilage degradation through the induction of oxidative stress within the joint. Studies by Steinbeck, et al. showed that in early OA patients, synovial macrophages produced reactive oxygen species (ROS), hypochlorous acid, chlorine gas and chlorinated peptides, which exert an oxidative effect on cartilage [101]. Consistently, myeloperoxidase, an enzyme responsible for the production of chlorine gas, hypochlorous acid and chlorinated peptides, appeared elevated in early OA compared to controls and end-stage OA [101]. Furthermore, a high level of iNOS, which induces nitric oxide (NO) production, has been detected in OA patients [38]. Specifically, double immunohistochemical staining showed strong iNOS expression in CD68+ macrophages in the synovial lining and sublining layer of advanced OA patients [102]. The release of NO results in oxidative stress and impacted immune processes mediating cartilage integrity [102]. These findings were confirmed in a recent study, showing that NO production in synovial macrophages was significantly elevated in modest to severe knee OA patients compared to controls [103].
Next to cartilage degeneration, the MMP release by activated synovial macrophages also permits osteophyte formation and/or progression through cartilage matrix remodelling [104,105]. Osteophytes are bony outgrowths at the joint margin (Figure 1) and their presence is associated with pain in OA [12]. In early collagenase-induced OA, synovitis is considered a key contributor to osteophyte formation [106]. Synovial macrophages also release growth factors that can induce osteophyte formation, such as TGF-β [28] (Figure 1). Moreover, macrophage cell surface marker levels were directly proportional to osteophyte formation [25].
A study in collagenase-induced OA murine knee joints revealed that depletion of synovial macrophages resulted in an 85% reduction of osteophyte formation after 7 days [28]. This animal model shows resemblance with human knee OA and is claimed to be a suitable model for studying osteophyte formation during OA. Remarkably, in another mouse model of papain-induced OA treated with triamcinolone acetonide (TA), Siebelt, et al. reported that enhanced macrophage infiltration diminished osteophyte formation [66]. This effect might have been caused by increased proportions of CD163/FRβ-positive M2 anti-inflammatory macrophages, which, in in vitro cultures, displayed increased IL-10 mRNA expression [66]. These findings underline the potential importance of macrophage polarization in osteophyte formation in OA.
Osteophyte formation is regulated by growth factors produced by synovial macrophages, in particular TGF-β [28]. Van Lent, et al. showed that macrophages induce osteophyte formation through the production of TGF-β and by the expression of factors that induce chondrogenesis of mesenchymal cells [107]. Furthermore, their studies highlighted the possible crucial role of bone morphogenic proteins (BMPs) in osteophyte formation. Additionally, in patients with advanced OA, hepatocyte growth factor (HGF) stimulated the macrophage-selective production of TGF-β1 and BMP-2 [76]. These findings pinpoint TGF-β and BMP as key players in osteophyte formation in OA, even though macrophages might not be the exclusive producers of these growth factors [28].
In addition to growth factor production, the role of the alarmin proteins S100A8/A9 in osteophyte formation has been studied in collagenase-induced OA, DMM-induced OA and in patients with early OA [104,105]. Alarmins are released by activated macrophages and signal through TLR-4 [106,108]. Elevated alarmin S100A8/A9 plasma levels in patients with early knee OA correlated with development of osteophytes over 2 and 5 years [104]. Further research is required to uncover a possible mechanistic relationship between alarmin secretion and osteophyte formation.
In OA, subchondral bone remodeling is spatially variable and related to disease stage [109]. Bone resorption is increased in early stages, whereas in advanced stages ectopic bone formation predominates [109]. Macrophages can influence this process of bone remodeling, which involves bone resorption by osteoclasts and formation by osteoblasts [109]. The multinucleated osteoclasts originate from the same precursor cells as macrophages through stimulation of M-CSF and receptor activator of NF-κB ligand (RANKL) [110,111]. Moreover, macrophages are able to induce osteoclastogenesis though the production of inflammatory cytokines, including IL-1, TNF and IL-6 [110]. These cytokines promote osteoclastogenesis both directly by stimulating osteoclast precursors and indirectly by inducing RANKL on synovial fibroblasts [110,111].
Several studies have shown that macrophages isolated from the synovial fluid of knee OA patients differentiated into osteoclasts and showed lacunar resorption upon RANKL stimulation [112-114]. However, Adamopoulos, et al. reported greater osteoclast differentiation upon RANKL stimulation of RA synovial fluid macrophages as compared to OA synovial fluid macrophages [112,113]. Nevertheless, although osteoclast differentiation of synovial fluid macrophages may be more relevant in RA, the induction of osteoclast formation by macrophages could still be a contributing mechanism involved in, for instance, osteophyte formation in OA.
Synovitis has been recognized as one potential determinant of pain in OA patients and in animal models [2,38,77,115,116]. Synovitis as observed by MRI is related to knee pain [117], even more when using contrast-enhanced MRI [118,119] or dynamic contrast-enhanced MRI [120]. However, another study reported that changes in synovitis were not associated with concomitant changes in pain [96]. Also, in an animal model of collagenase-induced OA, the peak of synovitis did not correlate with the detection of pain [121]. Whether these contradictory findings relate to different OA phenotypes in patients and/or are merely limitations of animal OA models, warrants further investigation [116].
The specific association between macrophages and OA pain has been described in a monosodium iodoacetate (MIA) rat model of OA [116]. Furthermore, macrophage infiltration correlated with pain in patients with varying degrees of human knee OA [29]. These infiltrating macrophages were activated based on shedding of CD163 and CD14 markers and were associated with pain [29]. Another study confirmed the association of macrophage CD14 expression with pain in patients with knee OA [25]. Furthermore, elevated expression of CCL2 in synovium was found in patients with symptomatic OA and CCL2 level in the synovial fluid of these patients was associated with pain [85]. Conceivably, these mediators play a role in OA-related pain through the attraction of macrophages and other immune cells, resulting in an augmented immune cell infiltrate [115].
Apart from macrophage infiltration, several studies have demonstrated that cytokine release by macrophages is also related to pain in OA. Notably, serum levels of TNF-α were associated with pain in patients with symptomatic OA [122], whereas in severe OA, IL-6 release correlated with pain [123]. IL-6 can be secreted by either macrophages or T cells, but the cellular source of IL-6 in OA is unknown [123]. Synovial fluid levels of macrophage migration inhibitory factor (MIF), a pro-inflammatory cytokine mainly produced by macrophages, was positively correlated with pain in a study of patients with Kellgren-Lawrence (KL) grade 2-4 knee OA [124]. MIF induces pro-inflammatory cytokine release by macrophages, which might explain this correlation. Finally, synovial macrophages displayed increased nerve growth factor (NGF) expression, which was associated with pain in OA [77]. Together, these findings underscore a positive correlation between soluble macrophage mediators and pain in knee OA.
Nevertheless, preliminary findings of a study in end-stage knee OA demonstrated that higher levels of GM-CSF and its receptor CD116 were correlated with reduced pain, independently of the expression of other macrophage markers [125]. This seems counterintuitive since GM-CSF and its receptor are involved in macrophage survival and attraction [126]. However, in early collagenase-induced OA, pain was reported to be GM-CSF dependent [126]. Given the fact that stimulation of monocytic progenitor cells might be more pronounced in early disease, this could explain the observed differences between early and advanced OA.
Studies by Klein-Wieringa, et al. and De Jong, et al. demonstrate that presence of synovial CD4+ T cells rather than macrophages is associated with knee pain in advanced OA [45,46]. Nevertheless, an indirect role of macrophages may still be envisioned as they are involved in activation of CD4+ T cells and T cells activate macrophages in return.
Glucocorticoids slightly reduced CD68+ macrophages in the lining layer in patients with symptomatic knee OA, but this did not translate into different MCP-1, MIP-1 alpha, MMP-1, MMP-3, TIMP-1, and TIMP-2 levels in the synovial lining and sublining layers [127]. Rochetti, et al. reported a reduction of macrophage numbers in advanced knee OA after intra-articular injection of hyaluronan or methylprednisolone [128]. Mechanistically, this involved two processes; hyaluronan appeared to mainly stimulate reparative processes, whereas the corticosteroid appeared to reduce the inflammatory process [128].
Intra-articular therapies might not only reduce macrophage numbers, but also modulate macrophage phenotypes. Glucocorticoids increase synovial macrophage expression of CD163, a proposed marker of M2 macrophages producing IL-10 [25,62,65,66]. Utomo, et al. added dexamethasone to synovium explants of OA patients and showed an anti-inflammatory effect in gene expression analyses [62]. In addition, dexamethasone was added to primary human monocytes that were first polarized in vitro in either M1 or M2 phenotypes. Dexamethasone suppressed the pro-inflammatory M1 macrophages and enhanced the anti-inflammatory M2 macrophages. Similar experiments were performed with rapamycin, bone morphogenetic protein 7 (BMP-7) and pravastatin. Generally, rapamycin and BMP-7 enhanced the inflammatory response in synovium explants and suppressed M2 macrophages. Pravastatin had no impact on the inflammatory status of the explants but did suppress M2 macrophages. Furthermore, because of different results between untreated synovium explants and explants that were pre-treated with IFN-γ and TNF-α, it was concluded that the impact of the tested compounds may be different between disease severity levels [62].
In a rat model of papain-induced OA, intra-articular triamcinolone acetonide (TA) injections resulted in reduced osteophyte formation but did not affect cartilage degeneration or subchondral sclerosis. Mechanistically, in vitro experiments showed that TA elicited its effect by inducing monocyte differentiation to M2 macrophages [66].
A currently investigated approach aiming at macrophage skewing is Tissuegene-C, a cell-mediated gene therapy modality for localized delivery of TGF-β1. In a rat MIA model, IL-10 production and other M2 macrophage markers were elevated in knee joints of the Tissuegene-C group as compared to the control group, although M1 macrophage numbers were similar. These findings suggest that Tissuegene-C induces an anti-inflammatory environment in the knee joint [130]. Tissuegene-C is currently being tested in phase II clinical trials in knee OA patients [129].
So far, targeting cytokines has yielded disappointing results [7]. For instance, anti-NGF-β therapy in knee OA patients resulted in substantial pain reduction but was accompanied by serious side effects [7]. Furthermore, anti-IL-1β treatment has failed to improve clinical outcomes in OA [7], even though IL-1 is a key cytokine produced by activated macrophages. Finally, anti-TNF therapy has shown similar, disappointing results in OA [7].
The lack of response in OA patients may be a result of their heterogeneity. Schue, et al. compared high and low cellularity subgroups of radiographic knee OA with respect to response to anti-TNF treatment with infliximab. At baseline, the high cellularity subgroup showed increased numbers of mononuclear cells and blood vessels and higher COX-2 and IL-1 expression compared with the low cellularity subgroup, whereas other cytokine levels were similar. Remarkably, only patients with high synovial cellular infiltrate responded to anti-TNF treatment although groups were small [130]. This finding highlights the heterogeneity between patients and its potential importance for the response to treatment.
Moderate, positive effects were observed by broadly-acting anti-inflammatory drugs. For example, in ACLT rabbits, statins inhibited CCL2 and MMPs, reduced infiltration of CD68+ macrophages in the OA subintima [99] and decreased articular cartilage degradation. Likewise, CCL9 neutralization reduced macrophage and CD4+ T cell infiltration and pro-inflammatory IL-1β expression in ACLT mice, which translated into less severe histopathological signs of OA, decreased osteoclast formation, and decreased MMP-13 expression [131]. Furthermore, a study in patients with advanced knee OA revealed that celecoxib treatment decreased macrophage infiltration and cytokine expression in the synovial membrane, independent of cyclooxygenase (COX)-2 inhibition [132].
Methotrexate (MTX) may exert a positive effect on OA as the folate receptor β on synovial macrophages may serve as an entry route [71]. A pragmatic phase III trial of MTX in knee OA patients is currently running [133]. However, given the lower number of FRβ positive macrophages in OA as compared to RA, its efficacy might be less in OA [42]. To this end, assessment of FRβ expression by non-invasive macrophage imaging [25,29] could prove a useful diagnostic tool to identify OA patients eligible for MTX therapy.
Synovial macrophages are thought to be key players in OA and appear to play a role in the characteristic osteophyte formation, cartilage degeneration and pain. Several studies have suggested that macrophages in OA synovium are the main producers of inflammatory mediators, which in turn induce release of proteinases, aggrecans and growth factors by synovial fibroblasts. Although activators of synovial macrophages remain unknown, it is hypothesized that ECM debris activates macrophages in OA, which in turn aggravates the anabolic and catabolic imbalance in the OA joint. Therefore, synovial macrophages in synovitis are considered as critical drivers of OA.
In synovitis, synovial macrophages are mostly present in the lining layer of the synovium and irregularly distributed in the synovial sublining layer. These macrophages display both M1 and M2 phenotypes, as evidenced by expression of cell surface markers, gene expression, and cytokine profiles. Although the M1/M2 dichotomy is a highly simplified concept and mainly based on in vitro observations, it is a helpful tool. Unfortunately, in most OA studies, macrophage polarization is not fully characterized. Nevertheless, it has become clear that the polarization state of synovial macrophages varies between OA patients and between disease severity stages. More research is warranted to gain insight in the role of distinct macrophage phenotypes.
The role of macrophages might differ in early and advanced OA. It is hypothesized that innate immunity plays a key role in early OA pathology, whereas in advanced stages adaptive immunity is more prominent. However, macrophages attribute to both immune responses. Macrophages have mainly been studied in animal models, ranging between DMM, ACLT, collagenase-induced and spontaneous OA models. However, differences in models should be noted and cautious interpretation of results is warranted. The surgically-induced DMM model of OA is not associated with inflammation, although minor inflammation can be triggered by the surgical procedure, whereas the collagenase-induced model is associated with significant synovitis [50]. As a result, different models may draw different conclusions about the role of macrophages in OA.
To conclude, as synovial macrophages are considered key drivers of synovitis and are associated with OA progression, targeting synovial macrophages represents a potential treatment approach. Therapies involved in macrophage phenotype modulation are promising; however, more knowledge is required about macrophage polarization in different disease stages and between OA phenotypes. Synovial macrophage imaging and marker characterization might be able to identify knee OA patients eligible for macrophage-targeted therapies. Hence, future research should focus on the role of macrophages in early and advanced OA and between different phenotypes of OA patients.