Long considered as orphan diseases because of their poorly estimated frequency. Genodermatoses are more and more encountered in the world. They have been reported in all ethnic groups, including African blacks. In Mali, ethnic diversity and the frequency of consanguineous marriage justify the interest of this study.
To determine the prevalence of genodermatoses in Mali, to describe their clinical and therapeutic aspects.
It was a descriptive cross-sectional study of all cases of genodermatoses diagnosed and followed in the dermatology department of the Centre National d'Appui à la lutte contre la Maladie de Bamako National support center for disease control (CNAM) of Bamako, regardless of their age and sex, during the period January 2015 to December 2018.
A total of 153 cases followed for Genodermatoses out of 4372 consulted were included in our study, ie an overall prevalence of 3.5%. Albinism accounted for 48.36% (74 patients), Congenital Ichthyosis 14.37% (22 patients), Hereditary Epidermolysis Bullosa 10.45 (16 patients), Neurofibromatosis 11.76% (18 patients), Xeroderma Pigmentosum 9.16% (14 patients), Incontinentia Pigmenti 5.88% (9 patients). Females accounted for 54.12% (83 patients), males accounted for 44.88% (70 patients) and children 41.83%. Clinically, hyperpigmented macules, actinic keratoses, and photosensitivity were observed in Albino and Xeroderma Pigmatosum. Hereditary Ichthyosis was characterized by cutaneous dryness associated with squamous lesions in 95% of cases. All evolutionary stages of L'Incontinentia Pigmenti were observed in our study. Therapeutically, all our patients have received adequate care with regular follow-up. Hereditary epidermolysis bullosa received local treatment with twice-daily baths with dermatological or antiseptic soaps, the use of topical antibiotics on erosive lesions. Oral antibiotics were reserved for cases of proven superinfection. Pigmented spots and actinic keratoses were treated by the application of liquid nitrogen. Sunscreen creams were routinely used in all albinos and Xeroderma Pigmentosumen in addition to advice on the importance of sunscreening. The 6 cases of carcinoma were treated surgically.
Genodermatoses have the same clinical manifestations as those observed in Western countries, the prevalence seems to be different in sub-Saharan Africa favored by inbred marriage.
Genodermatosis, Epidemiology, Clinical, Therapeutic, Mali
Genodermatososes are genetic diseases of the skin: A group of inherited disorders with a conglomeration of cutaneous and systemic signs and symptoms. Long regarded as orphan diseases because of their frequency, poorly estimated by the absence of multicenter epidemiological studies, Genodermatoses are more and more encountered in the world. They include a heterogeneous group of chronic, rare, disabling or even lethal diseases due to a genetic mutation [1]. While some have an autosomal dominant inheritance such as Hereditary Bullous Epidermolysis (EBH), Incontinentia Pigmenti (IP), other Neurofibromatosis have an autosomal recessive inheritance such as Xeroderma Pigmentosum (XP), Oculocutaneous Albinism, Congenital Ichthyosis. Genodermatoses remain a major health problem and constitute a considerable burden, both for the child and for his family: disability and limited life expectancy [1,2]. Genodermatoses seem to be a cosmopolitan disease, they have been reported in all ethnic groups, including African blacks. In the world there are between 3,000 and 4,000 cases of Genodermatoses but there are still many unidentified patients [2]. Their epidemiological profile differs from one entity to another and from one continent to another. Many studies have questioned the notion of parental consanguinity in children with these genodermatoses. Chronic diseases, most have a carcinogenic evolution. The diagnosis is essentially clinical based on careful clinical examination including interrogation and dermatological examination. Biology, histology and genetics, in addition to confirming the diagnosis, are of paramount importance in research. The treatment is generally symptomatic and the prevention of the appearance of cutaneous and precancerous lesions proves essential for the follow-up of these children. At present, etiological treatment is the subject of numerous studies [1,3,4]. The heterogeneity of the clinical expression, the tumoral risks and the dangerous prognosis of this disease impose a regular follow-up of the subjects affected by these genodermatoses [3,5,6]. Given the rarity of genodermatoses in developed countries, it is important to study the epidemiological and clinical characteristics of this pathologies in order to have a comprehensive view. In general, very few studies or even absent, associated with genodermatoses were carried out in the Malian population. Ethnic diversity and the frequency of consanguineous marriage of Mali justify the interest of the present study, whose purpose is to determine the frequency of Genodermatoses, to describe their clinical and therapeutic aspects.
This is a hospital-based observational study carried out on a series of 153 genodermatoses cases, aged 2 months to 66 years. and diagnosed ed at the dermatology department of the << Centre National d'Appui pour la lutte des Maladies (CNAM) >>, over a period of 4 years from January 2015 to December 2018. CNAM is located in the center of Bamako, Mali. it is one of the reference structures of the country for the management of skin diseases with within it a unit for the care and monitoring of genetic diseases. Any male or female patient diagnosed with genodermatosis, the availability of socio-demographic and clinical characteristics, acceptance to sign a consent or assent informed were considered as criteria for inclusion. A free consent of all parents of children were obtained before their inclusion. After obtaining a signed informed consent/assessment for each participant, information on age, gender, genodermatoses subgroup phenotypes including albinism, congenital ichthyosis, hereditary epidermolysis bullosaa, neurofibromatosis, Xeroderma pigmentosum and incontinentia, and treatment were collected. Routine biological (blood count, total leukocyte count, differential leukocyte count, erythrocyte sedimentation rate, liver function and renal function tests), urine, microscopic and radiological examinations were performed in all participants. Incomplete medical records, non-signing of consent/assent were excluded from our study.
EPI INFO software version 6.04 was used for statistical analysis. A pre-established Excel file was done to collect the socio-demographic and clinical characteristics of the patients. Absolute numbers and percentages were calculated from categorical variables. The figures (histogram and Pie chart) were produced using Microsoft Excel software.
A total of 153 cases followed for genodermatoses out of 4372 consulted were included in our study with an overall frequency of 3.5%. The age mean was 37.33 ± 4.41-years-old (Table 1). In the present study, females were over-represented (83.54%) than males (70.46%) Figure 1.
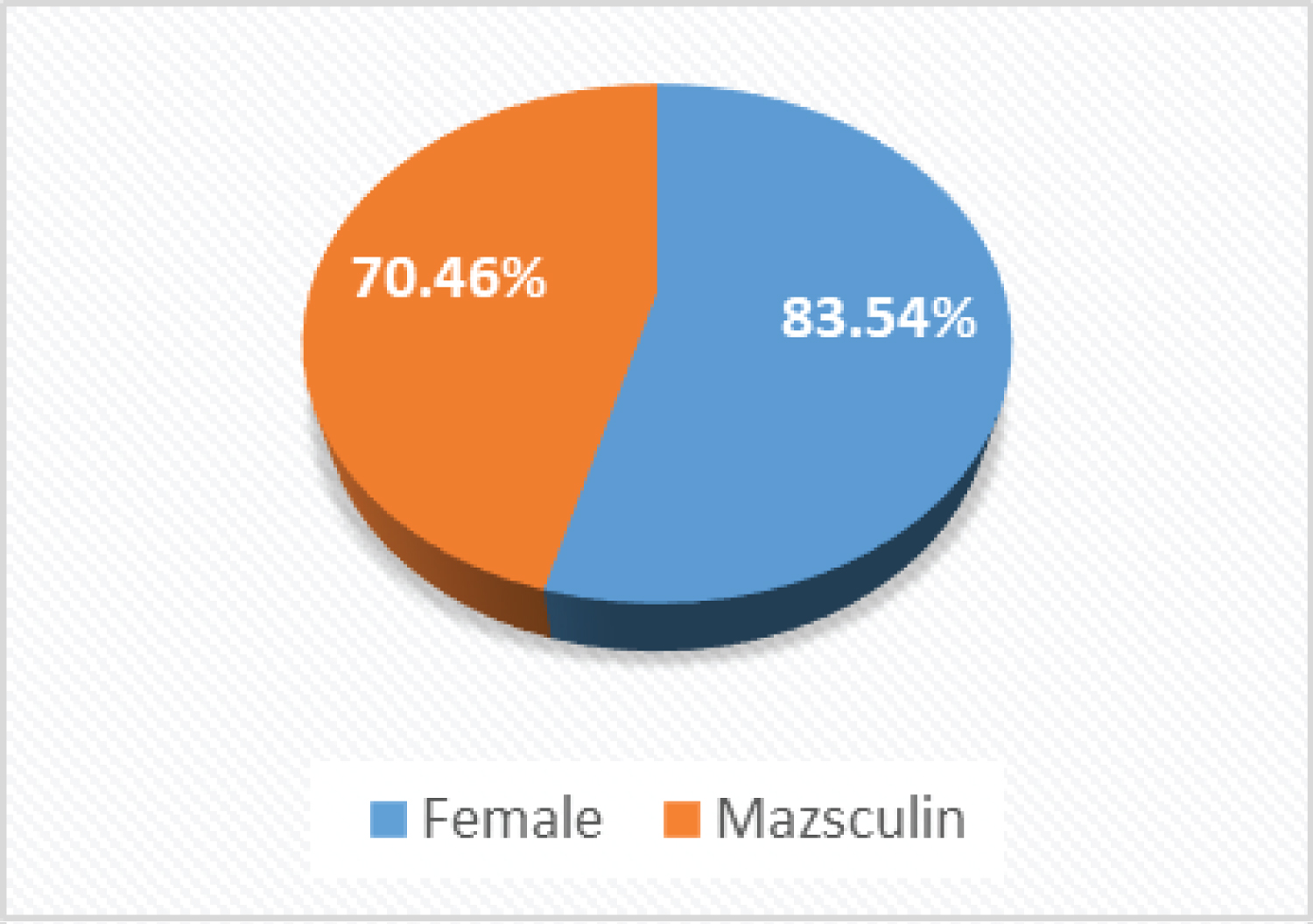 Figure 1 : Distribution of the cases by gender.
View Figure 1
Figure 1 : Distribution of the cases by gender.
View Figure 1
Table 1: Summary of essential data. View Table 1
Albinism was predominantly represented (48.36%) followed by Congenital Ichthyosis (14.37%), Hereditary Epidermolysis Bullosa (10.45%), Neurofibromatosis (11.76%), Xeroderma Pigmentosum (16%), Incontinentia Pigmenti (5.88%).
Table 2 shows the distribution of genodermatosis subgroup (Figure 2).
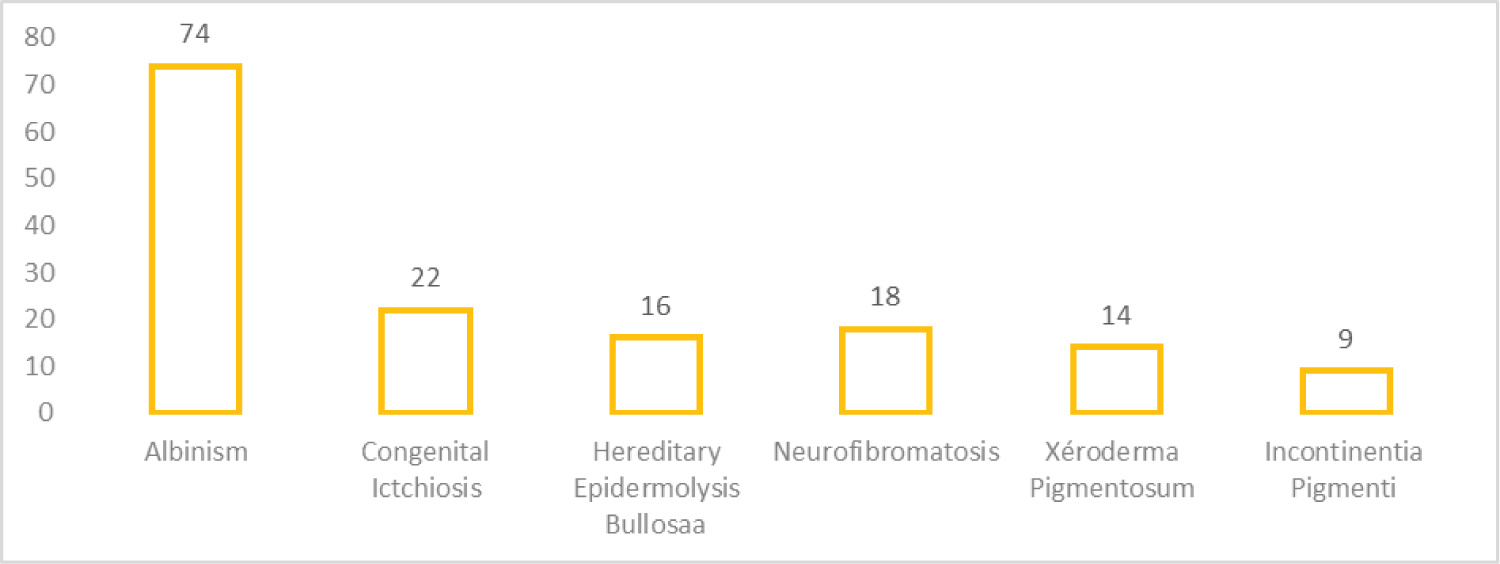 Figure 2: Distribution of Genodermatoses by type.
View Figure 2
Figure 2: Distribution of Genodermatoses by type.
View Figure 2
Table 2: Distribution of cases according genodermatosis subgroup. View Table 2
Clinically, pigmented spots and actinic keratoses were observed in all albino patients during their follow-up (Figure 3). 6 Albinos presented histologically of ulcero-budding lesions for a squamous cell carcinoma (Figure 3).
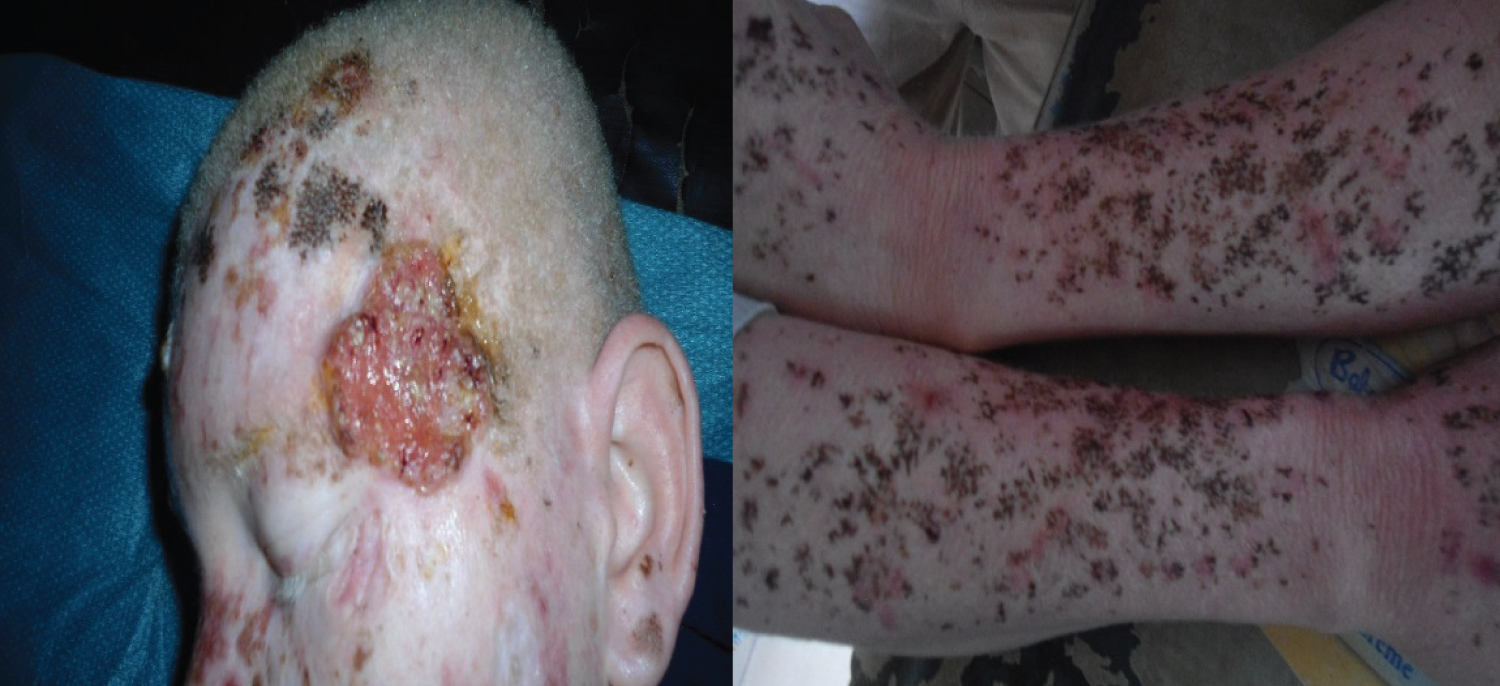 Figure 3: Carcinome épidermoide chez un albinos.
View Figure 3
Figure 3: Carcinome épidermoide chez un albinos.
View Figure 3
In Xeroderma Pigmatosum cases, in addition to erythematous and pigmented macules diffused throughout the integument, photophobia was observed in more than 70% of patients (Figure 4). In the Incontinentia Pigmenti, stage 3 with these macular hyperpigmentations, of variable color, very specific disposition and distribution and following the lines of Blaschko was observed in 45.20% of cases, stage 4 with achromatic macular lesions, atrophic depigmented, alopecic, often linear in 32.10% of cases and stage 2 with verrucous, hyperkeratotic, sometimes hypertrophic and lichenoid lesions arranged blachko-linearly in 11.30% of cases and stage 1 with vesicular lesions linear tumblers in 11.30% of cases. Hereditary epidermolysis bullosa presented cyclical erythematous-vesiculobullous lesions and erosions of variable localization in 85% of cases. Histological examination classified these EBH cases as simple EBH in 42.40% of cases, Junctional EBH in 22.10% of cases and Dystrophic EBH in 35.50% of cases. For the cases of Neurofibromatosis, Lentigine was observed in 40.20, the spots Coffee with milk in 67.40% and Neurofibromas in 80.70% (Figure 5 and Figure 6). These lesions were found in all our patients and these lesions were localized with the Trunk 26.35% Lower member 23.72% Upper member 21.16%, Cephalic 18.44% and Neck 10.5%. The bottom of the eye and the electroencephalogram were systematically requested in all our patients and these results were normal in 72.8% of cases. We did not have any cases of seizure. Hereditary Ichthyosis was clinically manifested by squamous lesions with thickening of the skin. Three (3) cases of erythroderma on erythrosis were observed. The concept of pruritus was observed in 76.32% of patients with Ichthyosis.
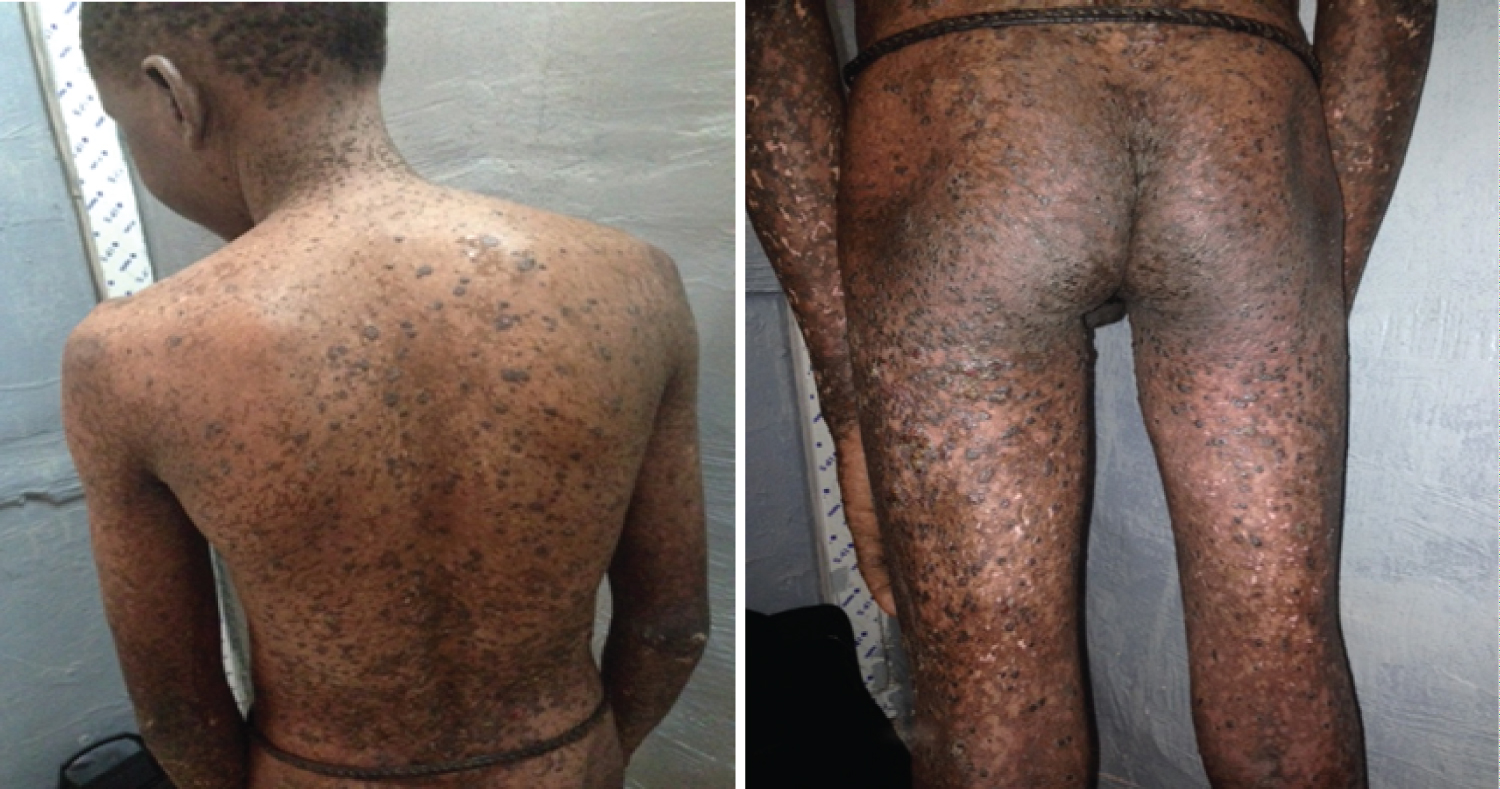 Figure 4: Macules hyperpigmentées de XP.
View Figure 4
Figure 4: Macules hyperpigmentées de XP.
View Figure 4
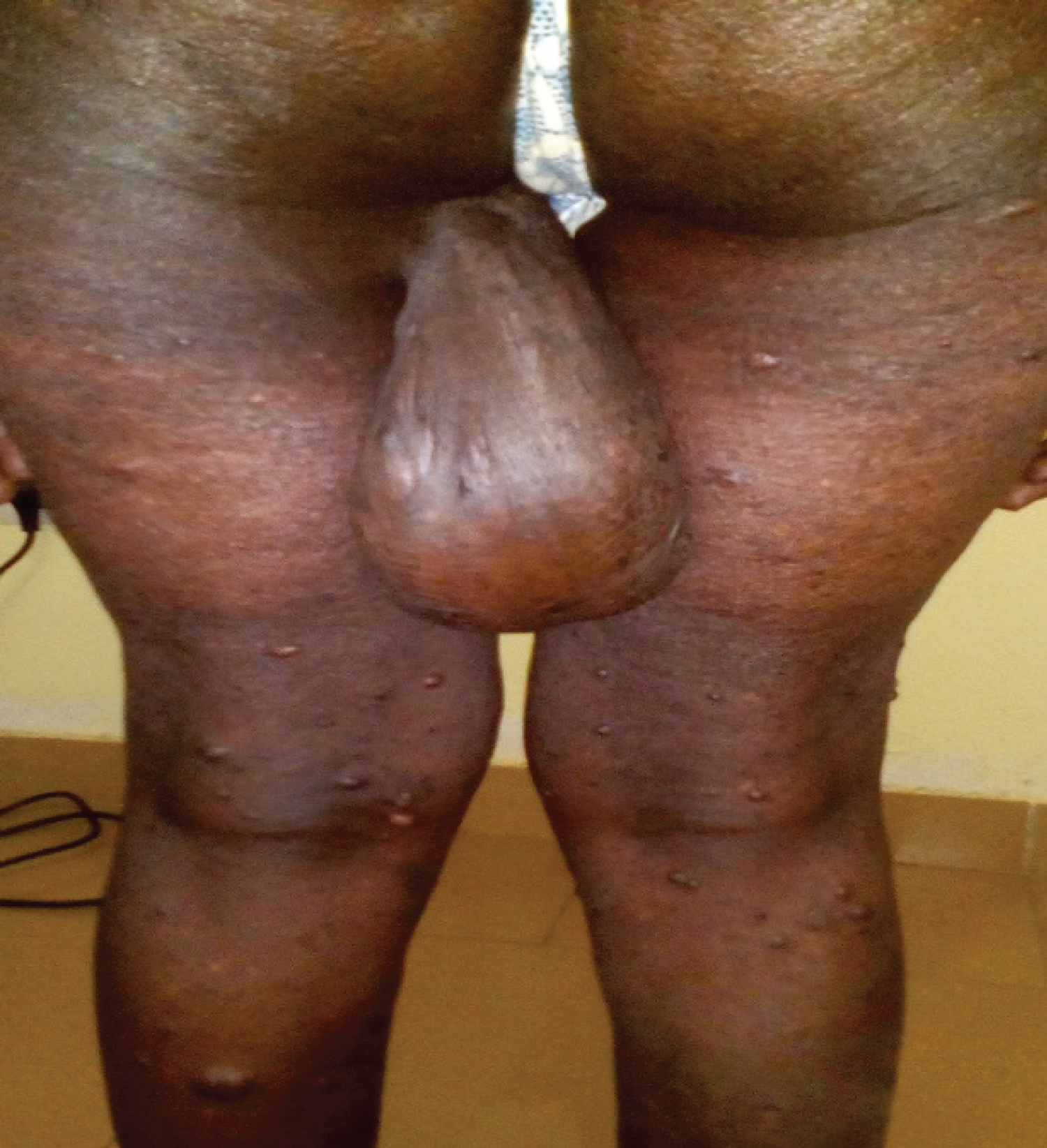 Figure 5: Multiple neurofibromas on the lower limbs.
View Figure 5
Figure 5: Multiple neurofibromas on the lower limbs.
View Figure 5
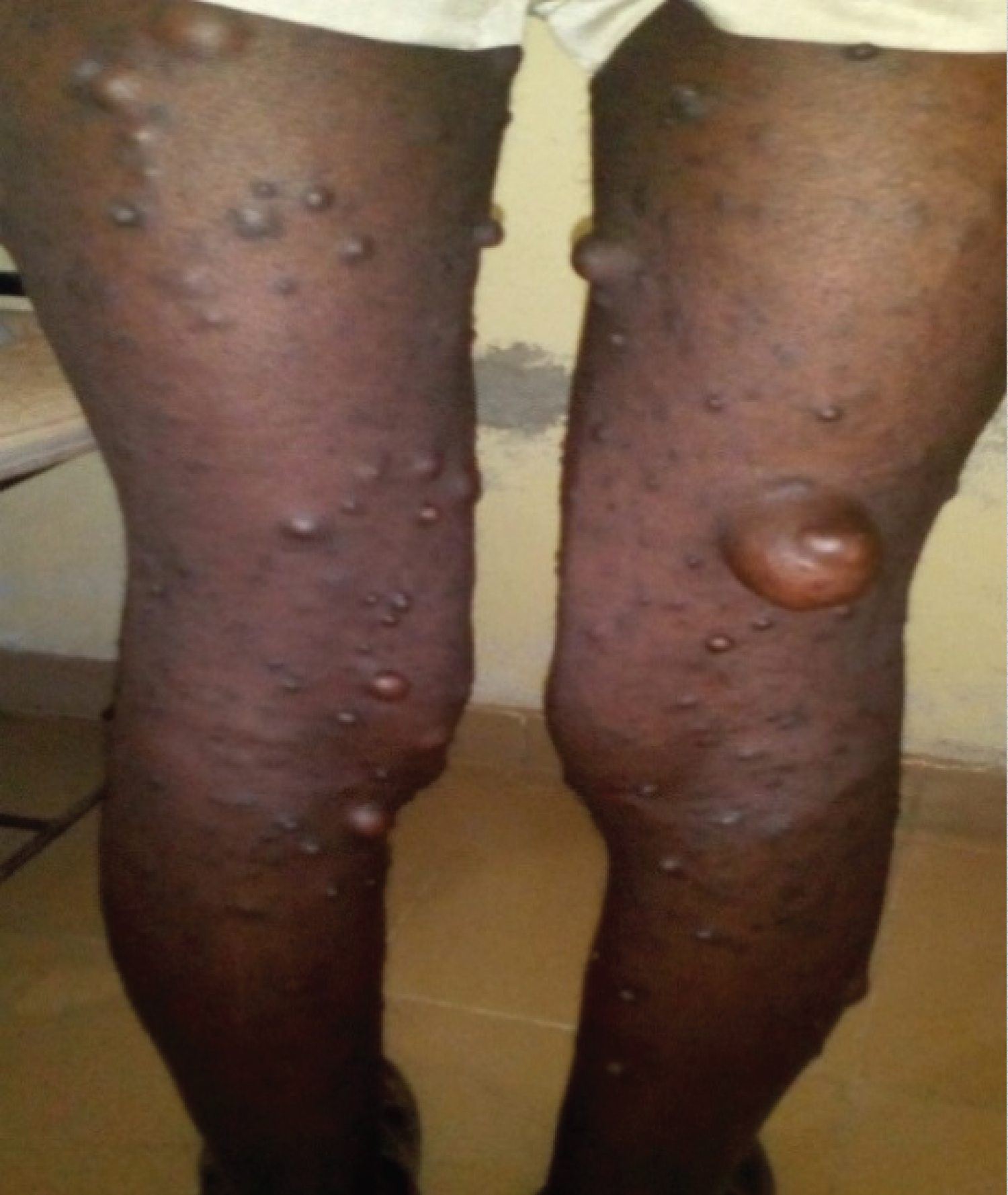 Figure 6: Royal tumor under gluteal with multiple small neurofibromas.
View Figure 6
Figure 6: Royal tumor under gluteal with multiple small neurofibromas.
View Figure 6
Therapeutically, all patients received adequate care with regular follow-up. Hereditary epidermolysis bullosa received local care to prevent the risk of infection to obtain a rapid regression of bubbles and erosions and finally to ensure the comfort of the child. These local treatments included a twice-daily bath with dermatological or antiseptic soaps, the use of topical antibiotics on erosive lesions. Oral antibiotics were reserved for cases of proven superinfection. Hereditary Ichthyoses were treated with emollients (5% vaseline and 10% urea) and Keratolytics (salicylic acid in petrolatum). For neurofibromatosis, the therapeutic abstention associated with the councils was of rule but some neurofibromas cases of large sizes benefited from surgical removal. The albino pigmented spots and actinic keratoses were treated by the application of liquid nitrogen. Sunscreen creams were systematically used in all albinos, in addition to advice on the importance of sartorial protection. Six carcinoma cases were treated surgically. The sunscreen protection, the use of sunscreens, topical retinoids were recommended in Xeroderma Pigmentosum. Topical antibiotics (fucidic acid cream) were used on ulcerative-crustal lesions.
Genodermatoses are genetic diseases with skin manifestations. In light of our results, the relative frequency of genodermatoses cases during four year at the dermatology department was 3.5% (Figure 2). This result is higher than that found by Federica and al who found a frequency of 0.40% according to a study carried out in Italia at Ethiopians patients [7]. This could be explained by the fact Federica's study took place in a European country outside Africa and another explanation could be consanguineous marriage, which is more frequent in our rural situation. It is also slightly higher than those reported by literature. Especially in Northern Ireland, the incidence would be 1, 4/1000000/year and prevalence of 32/1000000 [8,9].
In Scotland, the prevalence would be greater 49/1000000 [9]. In the United States, more than 50000 cases of genodermatoses are known [4]. The incidence would be 1/50000 births in Scandinavia and Croatia [10]. In our study, the female sex predominates. This female predominance could be explained by unsightly appearence of the lesion, prompting these women to consult more than men. This result is comparable to that of Konaté I and al in Burkina faso who found a sex ratio of 0.80 in study conducted among albinos [11]. According the literatory, incomtinatia pigmenti is almost non-exixtent in the male subject because it letal in uter at the male sex. Being a genetic disease, genedermatoses can be observed in all ages with accentuated clinical manifestations in certain ages. The averall age in our study (37.33 years) could be explained by the fact that young people, especially the young girls, have an increased need to take care of their skin. Clinically, there is not a significant difference beetwen the genodermatosis observed in African countries and all other continents (Table 3). The difference is in the different phenotypes of the skin [12]. The risk of occurrence of skin cancer seems more frequent in Africa than in Westerners because of the strong sunshin, particularly in sub-saharan Africa countries where genodermatoses pose a major puplic health problem with difficulty in management. Oculocutaneous albinism and Xeroderma pigmentosum pose a risk of early onset of skin and mucosal cancers. The prevalence of mucocutaneous cancers in this group is estimated at about 75% [4,9,13]. In addition to the risk cancer, there is the risk of premature skin agingin the form of freckles and lentgo-like pigmentation.
Table 3: Mains clinical manifestations of genodermatoses. View Table 3
Neurofibromatosis 1 is associated with cognitive and motor deficits which this pathogenesis is not well understood [14]. Neurofibromas have a risk of malignant transformation of the neurofibroma into neurofibrosarcoma (type 1 only) in 53.42% of cases.
Therapeutically, the management of the genetic diseases is generally symptomatic with regular monitoring. Special emphasis must be placed on the occurrence of the skin and mucosal cancers by photoprotection and the early treatment of precancerous lesions. In our study, the albinos and Xeroderma pigmentosum benefited from photoprotection by sunscreen creams SPF50 and topical retinoids. The treatment of actinic keratosis, hyperpigmented spots was done by cryotherapy with liquid nitrogen. This therapeutic method was consistent with the data in the literature [2,4,10]. Hereditary ichthiosis patients received emollient creams and retinoids tablets (tablet per day). Patients with hereditary epidermolysis bullosa received local care, in order to prevent the risk of infection, to achieve rapid regression of bubbles and erosions, and to ensure the comfort of the child. These local treatments included twice daily bathing with dermatological or antiseptic soaps, the use of local antibiotics on erosive lesions. Oral antibiotics were reserved for cases of proven superinfection. This treatment protocol is similar to that of the CHU Saint André in Bordeaux, France. Surgery will remain the rule for the management of cutaneous cancers [4].
Genodermatoses have the same clinical manifestations as those observed in developed countries but the prevalence seems to be different in sub-Saharan Africa, favored by consanguineous marriage. A multinational study will be essential to determine the true prevalence of genodermatoses.