Alzheimer disease (AD), the most common form of dementia, is a heterogenous syndrome with various pathobiologically defined subtypes. The clinical diagnosis of probable AD is enabled by the recent ATN biomarker system, but the definite diagnosis is only possible at post-mortem according to the updated NIA-AA criteria. The recent developments in the clinical and neuropathological diagnosis of AD including its specific subtypes improving the evaluation of AD and its impact on public health are briefly discussed.
Alzheimer's disease diagnosis, ABC score, ATN biomarker system, Primary age-related tauopathy
Alzheimer disease (AD), the most common form of dementia that currently affects around 50 million people worldwide, was initially defined as a clinico-pathological entity. However, AD is a heterogenous, multifactorial continuum with several pathobiologically defined subtypes, currently referred to as Alzheimer clinical syndrome [1]. AD is diagnosed definitely at autopsy by the deposition of β-amyloid (Aβ) in extracellular plaques and in vasculature (cerebral amyloid angiopathy/CAA) and intraneuronal aggregation of abnormal hyperphosphorylated tau protein forming neurofibrillary tangles (NFT), associated by neuronal loss and cerebral atrophy, according to the updated NIA-AA "ABC" criteria [2]. This "ABC" score for AD neuropathological changes combines "A" for the phases of amyloid paques [3], "B" for the NFT stages [4], and "C" for the CERAD neuritic plaque score [5]. Non-demented patients usually show Aβ phases 1-3, Braak stages 0 to III/IV, and CAA stages 0-I, while dementia is usually related to Aβ stages 4-5, Braak stages V-VI, and CAA stages 2-3. Comparison of the mini-mental stage examination (MMSE) scores with neuritic NFT stages in 200 consecutive autopsy cases of elderly persons (mean age 81.4 ± 8.6 years), showed that non-demented persons (MMSE 26-30) had no or only minimal tau pathology, and severely demented, no longer testable persons (MMSE around 0) showed a cluster of high tau pathology (Braak stages V and VI), while mild to moderate dementia was associated with a wide range of Braak stages (Figure 1).
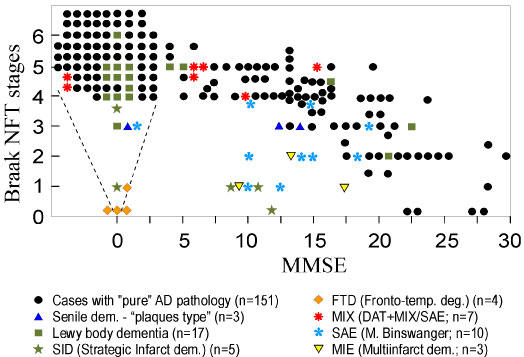 Figure 1: Relationship between Mini-Mental State Examination (MMSE) and Braak neuritic Alzheimer's disease stages in 200 consecutive autopsies of aged individuals (mean age at death 81.4 ± 8.6 years).
View Figure 1
Figure 1: Relationship between Mini-Mental State Examination (MMSE) and Braak neuritic Alzheimer's disease stages in 200 consecutive autopsies of aged individuals (mean age at death 81.4 ± 8.6 years).
View Figure 1
Diagnosis of probable AD in life is possible using the new ATN biomarker system [6]. It is based on relationships between markers of Aβ (A), tau pathology (T) and neurodegeneration (N). A and T have diagnostic specificity for AD, N is nonspecific for all neurodegenerative diseases. The neuropathological definition of AD drives the ATN definition of AD and requires the presence of amyloid and tau as evidenced by neuroimaging methods (PET) or cerebrospinal fluid (CSF) or plasma biomarkers for diagnosis, while N is shown by MRI, FDG PET or CSF total tau. A+, T+, and N+ cases have a worse prognosis than negative (A-T-N-) cases. The biomarker profiles and categories of the Alzheimer continuum referring to individuals with biomarker designation of either AD or Alzheimer pathologic change, are presented in Table 1. Prodromal AD is classified according to the ATN and NIA-AA criteria as follows: A-N- represents preclinical stage 0, A+N- preclinical stage 1, A+N+ preclinical stages 2 and 3, and A-N+ for suspected non-Alzheimer's pathophysiology (SNAP) [8]. The risk of progressive cognitive deterioration differs considerably between the various types: A-N- < A+N- < A+T-N+ (SNAP) < A+T+N+ [9].
Table 1: Biomarker profiles and categories (modified after [7]). View Table 1
During the last years, a new type of age-related dementia was described as primary age-related tauopathy (PART) [10]. It is morphologically characterized by tau pathology with Braak stages 0-IV, total or relative absence of amyloid (Aβ phases 0-2) and total absence of neuritic plaques. It predominantly involves people over age 80 years and is associated with mild to moderate cognitive impairment [11]. The relationship between PART and classical AD is under discussion, since the composition of NFTs between both types is identical, with both 3R and 4R isoforms. PART is considered either as prodromal form of AD or a subtype of AD-related disorders [12]. In PART, the lack of Aβ is responsible for less severe tau aggregation and, thus, for lower Braak stages and less cognitive impairment than in classical AD, since Aβ oligomers have been shown to potentiate tau aggregation by promoting tau seed uptake [13]. While classical AD increases from the 7th to the 9th decade and later shows mild decrease, the frequency of PART increases after age 85 years.
A comparative study of clinical and neuropathlogical diagnoses of AD in 3 epidemiologic samples reported a sensitivity of clinical or probable AD of 93% [14]. Meta-analysis of 20 (out of 1189) studies to distinguish autopsy-verified AD from other dementias or healthy controls showed a sensitivity of 85.4% (95% CI 80.9-90%) and a specificity of 77.7% (95% CI 70.2-85.1%). Values were higher for neuroimaging procedures and slightly lower for CSF biomarkers, while the combination of both resulted in higher values [15]. These data are related to two factors: (1) The various subtypes or variants of AD, and (2) The high frequency of co-morbidities in elderly people. The atypical variants of late and early onset AD are presented in Table 2. The three morphological subtypes are: Typical AD with tau pathology involving both cerebral cortex and hippocampus (74-82.5% of the total, mean age at disease onset 79 ± 4 years, at death 81 ± 9 years), the limbic-predominant type (tau pathology mainly involving the medio-temporal lobe (9-15% of total; mean age at onset 74 ± 6, at deat 85 ± 4 years), and the hippocampal sparing type (8-10.8% of the total; mean age at onset 68 ± 10, at death 76 ± 8 years) [17]. The duration of disease is shortest in the limbic-predominant type and longest in typical AD, which also shows more severe dementia than the two others [18]. These subtypes show specific clusters of the regional NFT densities [19], which is related to the regional vulnerability of neuronal subtypes to tau pathology, those in the entorhinal and other regions of the hippocampus showing early, basal forebrain mild, and neurons in cerebral cortex and locus ceruleus late stages of involvement [16]. Another major reason for the difficulties in clinical diagnosis of AD is the high frequency of co-morbidities or mixed pathologies in the aged brain. Table 3 shows the morphological diagnoses in a consecutive autopsy series of 1700 elderly demented patients (A) and those with clinical diagnosis of AD (B). AD-typical pathology was present in 82.9% of all demented people and in 92.8% of those with the clinical diagnosis of AD, but only half of them showed "pure" AD-pathology (ABC 3/3/3), while all the others were either atypical forms or showed additional cerebrovascular lesions, cerebral hemorrhages, Lewy pathology or other mixed pathologies, while vascular dementia in this cohort amounted for only 10.7 and 3.3% respectively. An earlier study of Alzheimer patients in clinical trials revealed pure Alzheimer pathology in only 31% and multiple pathologies in 63%, no pathological changes in 1% [20]. Similar co-morbidities in 1153 patients with pathological diagnosis of AD were shown recently [21]. This clinical and morphological heterogeneitiy of AD is due to multiple pathogenic factors - "upstream" genotypes causing co-morbidities, "downstream" disease-modifying gene variants (MPTP, H2 haplotype, APOE, TREM and GRN variants) - which induce misfolding of tau, Aβ, TDP 32, and others, resulting in various disease phenotypes [22].
Table 2: Atypical variants of late and early onset AD (modified after [16]). View Table 2
Table 3: Morphological diagnoses in consecutive Vienna autopsy series of A) demented and B) clinically diagnosed AD patients. View Table 3
In conclusion, AD is a multifactorial, heterogenous disorder, morphologically defined by the deposition of Aβ and hyperphosphorylated tau, that is definitely diagnosed at post-mortem according to the updated NIA-AA criteria, while diagnosis of probable AD in life is possible by the ATN biomarker system. The introduction of the ATN biomarker system has increased the accuracy of clinical diagnosis of AD compared to studies before general use of biomarkers including CSF and neuroimaging markers, such as β-amyloid and tau PET or multisystem neuroimaging methods. However, the validity of the current ATN biomarker system for the intra vitam diagnosis of AD and its differentiation from other dementia syndromes awaits further multicenter clinico-pathological studies using all currently available methods.
The still low diagnostic accuracy of the clinical diagnosis of AD despite the new ATN biomarker criteria is caused by the fact that the Alzheimer clinical syndrome includes several pathobiologically defined entities or subtypes which are clinically and morphologically different from each other:
• Alzheimer continuum (abnormal Aβ regardless of tau status)
• Alzheimer pathological changes (Aβ but normal tau - previously referred to as amyloid-predominant AD)
• Neurodegeneration but negative Aβ (SNAP)
• PART (abnormal tau but negative Aβ)
• Limbic predominant AD (tau pathology mainly involving the mediotemporal lobe - hippocampus)
• Hippocampal-sparing AD ("cortical sparing" of medial temporal lobes),
• Classical AD (abnormal Aβ and tau-pathology, the latter involving both cortex and hippocampus).
The prevalence of biological AD is greater than clinical probable AD at any age, in particular at age 85+ years [1]. These facts and the increasing incidence of AD illustrate its consequences on public health. In order to increase the sensitivit and specificity of the new ATN biomarker system, more extensive clinicopathological studies in well defined patient cohorts will be necessary, with post-mortem studies using the updated NIA-AA criteria. Similar proposals have recently been published for validating the criteria for the diagnosis of 4-repeat tauopathies [23]. Therefore, further interdisciplinary studies to improve our knowledge about the pathogenesis and to promote methods for early diagnosis of AD as basis for further preventive and successful therapeutic measures are urgently needed.
The author has no conflict of interest to declare.
The study was supported by the Society for Support of Research in Experimental Neurology, Vienna, Austria.