Elucidate the main clinical aspects of the CANOMAD spectrum.
Bibliographical review trough databases (PubMed, Google Scholar, Orphanet, Oxford Academic) of articles from 1985 [1] to 2019 and later selection of the most applicable of the above, in order to construct a non-systematic review.
CANOMAD is a chronic-ataxic autoimmune neuropathy associated with IgM monoclonal gammopathy. The correct diagnosis of this rare and multi-faceted disease will help optimal treatment.
CANOMAD, Chronic-ataxic neuropathy, Anti-disialosyl, IgM monoclonal gammopathy
CANOMAD is a rare syndrome that belongs to the group of peripheral neuropathies associated with IgM monoclonal gammopathy [2]. This acronym was first used in 1996 to describe the clinical features of the disease, which include: Chronic sensory ataxic neuropathy, ophtalmoplegia, M-protein with cold-agglutinin activity and anti-disialosyl antibodies [3].
The syndrome's physiopathology is not fully understood, but, according to the available researches, it will be expressed in patients whose serum contains IgM antibodies to disialylated gangliosides, mostly, with cold agglutinins demonstrated as well [4]. The high titles of anti-gangliosides will react against GD1b, GD3, GT1b e GQ1b, nerve cells elements, in the NeuNAc (alfa2-8) NeuNAc (alfa2-3) terminal epitope. This antibody-epitope binding occurring in sundry peripheral nerves structures, such as ganglion neurons of the dorsal root, peripheral axons and myelin, nodes of Ranvier, muscle spindles and motor nerve terminals will lead to neural involvement [5].
There are also presentations of cold agglutinin with anti-Pr specificity, which about half of patients with CANOMAD show, and it is associated to red cell activity of anti-Pr1 or anti-Pr2 [4]. Anti-Pr is the responsible for recognizing Pr antigenic determinant that is sialic-acid dependent and located in the red cell membrane. This justifies why IgM paraprotein with anti-Pr2 cold agglutinin activity may act identifying the sialic-acid epitopes present in disialylated gangliosides. Moreover, the anti-Pr IgM specificity might collaborate to the myelin binding by these antibodies, which also have pathogenic function in peripheral nerve demyelination, resulting in the CANOMAD affection [6,7]. In these patients, the worsening of neurological symptoms when the temperature gets low, may be explained by the presence of cold anti-Pr agglutinin may be justified by the anti-Pr cold agglutinin, as it causes cold agglutinin disease [6].
This disease mainly affects male patients with mean onset at 5th decade of life. The clinical presentation of this neuropathy is heterogeneous and the symptoms can appear in relapses [8]. The syndrome is typically manifested with proprioceptive ataxia, ophtalmoplegia and areflexia with relative preservation of motor function. In some cases, there is cranial nerve involvement with ocular, sensory or bulbar manifestations [2]. Motor weakness mainly affects oculomotor and bulbar muscles, therefore, CANOMAD is a chronic reminiscent of Miller Fisher syndrome [8].
Serologically, CANOMAD is characterized by the presence of IgM monoclonal gammopathy and antibodies against gangliosides with disialosyl groups, including GD3, GT1b, GD1b and GQ1b that share the terminal NeuAc(a2-3) Gal epitope in the glycide structure [9].
This nonsystematic review was conducted through articles in multiple databases, as PubMed, Google Scholar, Orphanet, Oxford Academic, applying key words as "CANOMAD", "IgM paraprotein", "chronic ataxic neuropathy", "ophtalmoplegia", "cold agglutinins", "anti-gangliosides", with and without the Boolean Operator "AND" between each pair of them. There were no exclusion factors regarding to idiom nor publication date. The analysis comprised since the pioneer study, in UK, 1996 by Willison and colleagues until the most recent, published in 2018.
This article aimed to elaborate a clear approach synthetizing the available information about the CANOMAD syndrome, it's physiopathology, clinical and serological manifestation as well as the disease's diagnosis and treatment based on the previous literature.
All articles accrued from the searches described, that were consistent with the proposed theme, were selected in order to secure a most reliable literature review. Naturally, those whose information about the matter was more valid, regarding the physiopathology, clinical manifestation, diagnosis and treatment of the CANOMAD syndrome, were included in this non-systematic review.
It is estimated that in 5-10% of the neuropathies, simultaneous serum paraprotein may occur. This justifies the importance of mentioning diseases that comprises both paraproteinemia and neuropathy, including CANOMAD syndrome, a rare phenotype that is associated to monoclonal gammopathy of undetermined significance (MGUS) [10].
In CANOMAD syndrome, the neuronal damage and further symptoms are caused when the GD1b, GD3, GT1b antibodies are recognized and connected to the respective oligosaccharide chain in peripheral nerves structures, such as peripheral axons, myelin, dorsal root ganglion neurons, nodes of Ranvier, and also motor nerve terminals and muscle spindles [4]. Thus, the IgM antiganglioside antibodies will deplete the gangliosides that are glycosphingolipids of the nerve cells membrane, causing degeneration of the sensory neurons by degrading its nodal axolemma and inducing demyelination. This process will result in CANOMAD [10,11].
There may also be cold agglutinins in CANOMAD's physiopathology, which include anti-Pr2 antibody nearby the PNS's capillary endothelia, framing an access of the immune mechanisms to PNS myelin through this brain-blood-barrier immune damage (Figure 1). Histologically, this syndrome will present either axonal degeneration or demyelination [1].
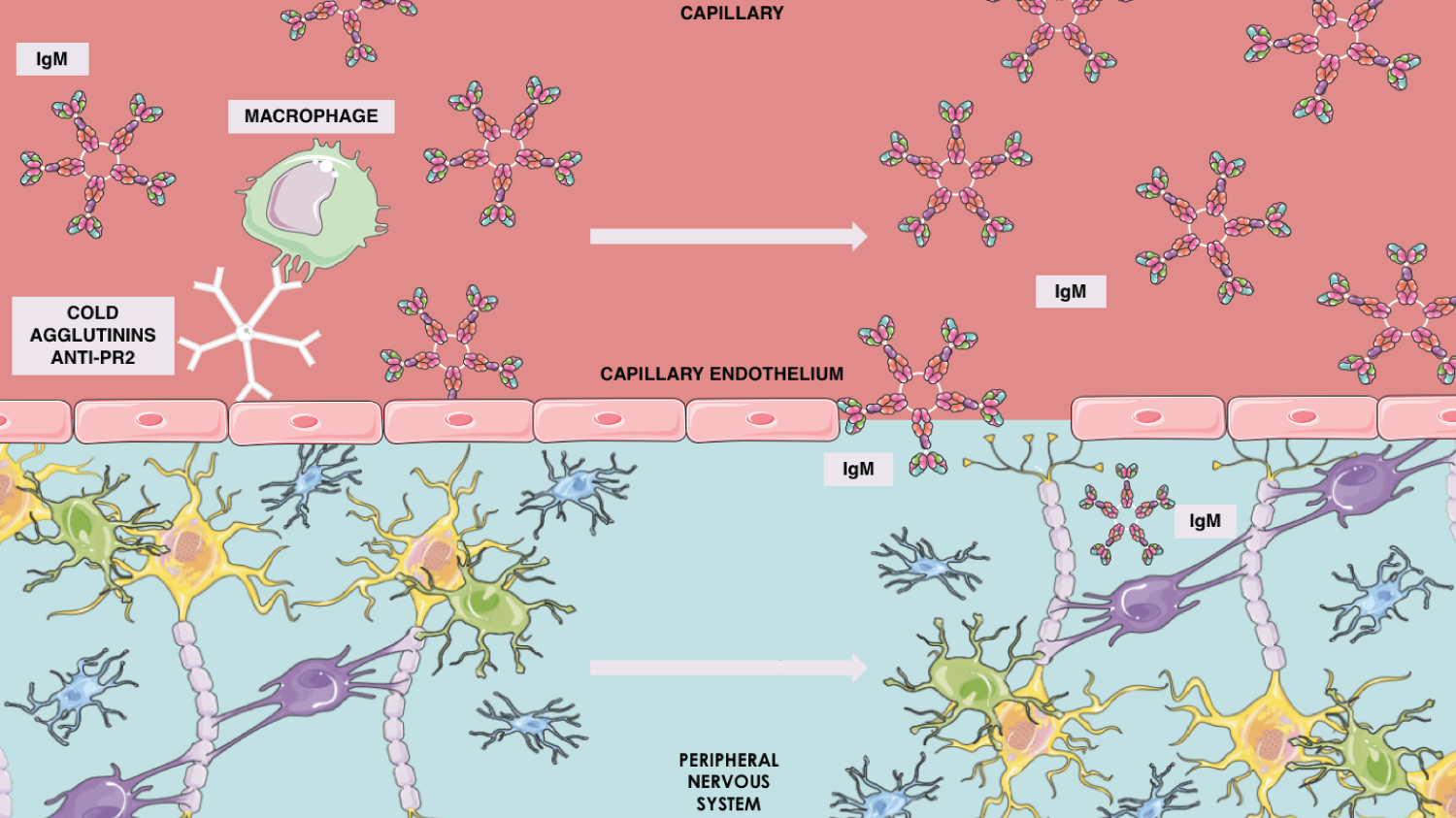 Figure 1: Anti-Pr2 cold agglutinin mechanism in IgM brain blood barrier penetration.
Figure 1: Anti-Pr2 cold agglutinin mechanism in IgM brain blood barrier penetration.
Illustrative elements from Servier Medical Art.
View Figure 1
Clinically, CANOMAD's onset is predominantly chronic, few are acute or subacute [12] and it affects mostly men around the age of 55. When this disease affects woman, its start is inclined to happen earlier, in the fourth decade [4]. Primarily, it comprises a chronic neuropathy with sensorial impairment and ataxic gait disorder accompanied by relatively preserved motor function in the limbs and areflexia or hyporeflexia in most cases. Sensory ataxia is found in all cases. The motor disturbances alter mainly oculomotor and bulbar muscles and may be either fixes symptoms or relapsing-remitting features. Also, cranial nerve sensory and motor involvement, fixed or relapsing-remitting, being the most affected the CN III, there from, the oculomotor commitment; it distinguishes CANOMAD from other paraproteinemic neuropathies [4]. Nevertheless, loss of kinesthesia, small fiber sensation, as numbness, vibration sense and proprioception, having relatively preserved muscle strength. Furthermore, acral and perioral paresthesia often featured as well [4].
Other manifestations are external ophthalmoplegia, dysphagia, dysarthria. Respiratory muscle weakness is rarely observed. Symptoms persistence is widely variable about its duration in years, from 4 years up to 4 decades, but often extended over decades [4,13]. It is reasonable to mention CANOMAD syndrome's course can be wavering over its chronicity [14].
Attesting CANOMAD syndrome is a challenge due to its rarity and miscellaneous symptoms criteria, besides some important differential diagnosis like Miller Fisher Syndrome and Guillain-Barré Syndrome, a wide investigation becomes necessary regarding to clinical and laboratory findings.
The diagnosis of CANOMAD requires having the clinical features of motor weakness, sensory ataxia, and in some cases reduced or absent reflexes as well as cranial nerve irregularities. Besides the presence of IgM antibodies against ganglioside with disialosyl groups, specificaly GD3, GT1b, GQ1b and GT1b. Antibody testing was perfomed via enzyme-like immunosorbent assay (ELISA) and the most patients had elevated serum IgM rates [10]. The presence of cold agglutinins varied according to the case. The clinical picture is variable as the antibody responds against desialylated gangliosides [8] (Figure 2).
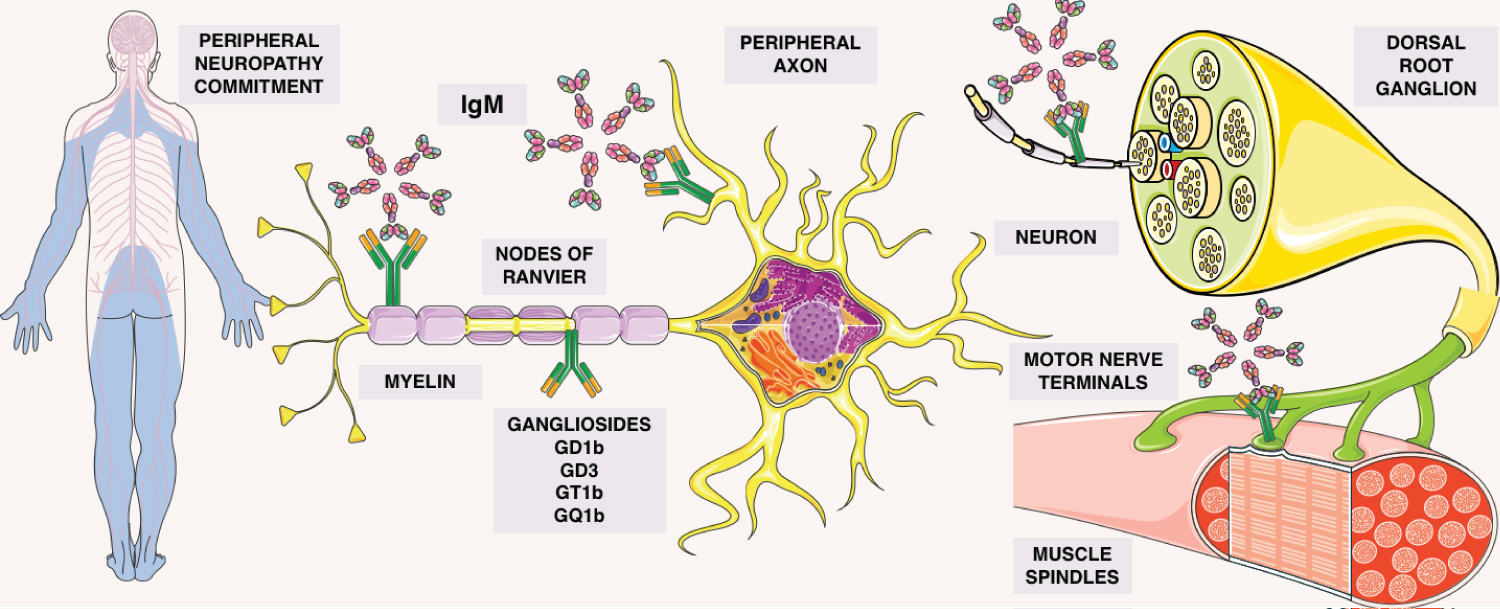 Figure 2: Description of the gammopathy involvement. Gangliosides attaching to motor nerve terminals, muscle spindles, nodes of ranvier, peripheral axons, myelin, in the dorsal root ganglions, allowing IgM adhesion in this sites. By autoimmunity, causing the commitment in this peripheral neuropathy.
Figure 2: Description of the gammopathy involvement. Gangliosides attaching to motor nerve terminals, muscle spindles, nodes of ranvier, peripheral axons, myelin, in the dorsal root ganglions, allowing IgM adhesion in this sites. By autoimmunity, causing the commitment in this peripheral neuropathy.
Illustrative elements from Servier Medical Art
View Figure 2
Neuroimaging exams (MRI and CT scan) usually is normal [2] but nerve biopsy indicate demyelination and axonal damage; also, electrophysiology and pathology show signs characteristic of axonal flawed [11]. The grading as severe, mildly deranged or normal is based in the demyelinating, axonal, motor and sensorial features [4].
Also, trace of nerve enlargement which is consonant with acquired demyelination was detected through neuromuscular ultrasound once performed in CANOMAD affected person. This finding was both qualitatively and quantitatively associated to demyelinating neuropathy in contrast to non polyneuropath patients. Eventually characterizing nerve ultrasound as a suggestion for additional diagnosis test, regarding the requirement of further analysis [1,14].
The nature of phenotype the CANOMAD is chronic, progressive and variable, what makes difficult to evaluate the effect of treatment [15,16]. Due to this and the scarce of case series, case reports and the absence of randomized trials, the treatment guideline is limited [10]. However, even if there is no evidence-based treatment, according to a few case reports, the therapy with intravenous immunoglobulin (IVIg) may be able to avoid fluctuations of symptoms in CANOMAD because of the antibody neutralization by IVIg improving nerve function [17].
Furthermore, the rituximab is considered to be optimal used in anti-MAG neuropathies. Yet, as a monoclonal antibody anti-CD20, it is not able to suppress CANOMAD activity, once the paraprotein IgM is antigen-induced by long-lived CD20 negative plasma cells, which explains the poor response to rituximab treatment [17,18]. Though, owing to the disease's site, rituximab might not be able to reach B Limphocytes in the nervous system due to the large size of the molecule incapable to cross the brain blood barrier [13,16].
CANOMAD is a chronic-ataxic autoimmune neuropathy associated with IgM monoclonal gammopathy. The correct diagnosis of this rare and multi-faceted disease will help optimal treatment.