Introduction: The urofacial (Ochoa) syndrome is a rare autosomal recessive disease characterized by congenital obstructive uropathy and abnormal facial expression.
Case presentation: We reported the case of a child aged six-years-old, the second child of consanguineous parents, the father reported that she has suffered from episodes of urinary tract infection, and enuresis. She was admitted in our department for an urinary tract infection. The clinical examination showed a characteristic, inverted, facial expression. The urea and creatinine were increased and GFR: 20 ml/min, renal echography showed an enlarged kidneys, with ureteral and calycial dilatation, and a low-compliance bladder with pseudodiverticulae, and a significant residual urine after voiding: 130 ml. Voiding cystourethrography showed a trabeculated bladder and a significant post-mictural residue.
The urofacial syndrome was suspected of: Facial characteristic expression + urinary abnormalities.
She was initially treated with antibiotic prophylaxis and anticholinergics, conservative treatment of renal failure and urinary self catheterization to void bladder.
Conclusion: Ochoa syndrome should always be considered in patients with dysfunctional bladder who have characteristic grimacing when smiling, The prognosis of urofacial syndrome is generally poor and requires multiple treatment modalities.
Ochoa syndrome, Characteristic grimacing, Dysfunctional bladder
The urofacial (Ochoa) syndrome (UFS) was first described in several unrelated Colombian families (Elejalde, 1979; Ochoa and Gorlin, 1987). As the name implies, UFS patients have both urinary and facial abnormalities. They present a bladder voiding dysfunction due to a lack of coordination between detrusor muscle contraction and urethral sphincter relaxation. This usually results in enuresis, urinary tract infection and hydronephrosis, leading to renal damage and eventually renal failure. The facial abnormality is a characteristic expression such that, when these patients smile, their facial musculature inverts and they appear to be crying. This grimacing is highly diagnostic of the UFS. In addition to facial and urinary abnormalities, constipation has been reported in about two thirds of the patients (Ochoa and Gorlin, 1987). The occurrence of UFS in multiple sibs with normal parents, the equal sex distribution, and the increased parental consanguinity indicate autosomal recessive inheritance for this condition. Using a combination of homozygosity-mapping and DNA pooling-strategies, Wang, et al. (1997) were able to place the UFS gene within a 1-cM interval on chromosome 10q23-q24. Mutation analysis has later excluded the glutamate oxaloacetate transaminase gene (GOT1), located in this region, as a candidate gene for UFS (Wang, et al. 1999). UFS is a rare condition of which the genetic background has recently been discovered. Mutations of HPSE2 and LRIG2 genes have been shown in these patients HPSE2 gene is encoding. The polypeptide heparanase-2 which is responsible in facial expression and urinary voiding control. LRIG2 encodes polypeptides which have role in neural signaling [1].
We report a Moroccan case of UFS, presented initially with acute renal failure.
This 6-year-old girl is the second child of consanguineous parents (uncle ± niece). Father is reported to have suffered episodes of urinary tract infection. He did not show the characteristic grimacing of his daughter.
This child was born at term following a normal vaginal delivery after an uneventful pregnancy. Birth weight was 2500 g and length 49.5 cm. Development was otherwise normal in the first years of life, but she suffered of enuresis. At the age of 6 she presented with urinary tract infection. The urea and creatinine were increased and GFR: 20 ml/min; renal echography showed an enlarged kidneys, with ureteral, and calycial dilatation, and a low-compliance bladder with pseudo-diverticular, and a signifcant residual urine after voiding: 130 ml. Voiding cystourethrography showed a trabeculated bladder and a significant post-mictural residue (Figure 1). This child showed the characteristic, inverted, facial expression (Figure 2).
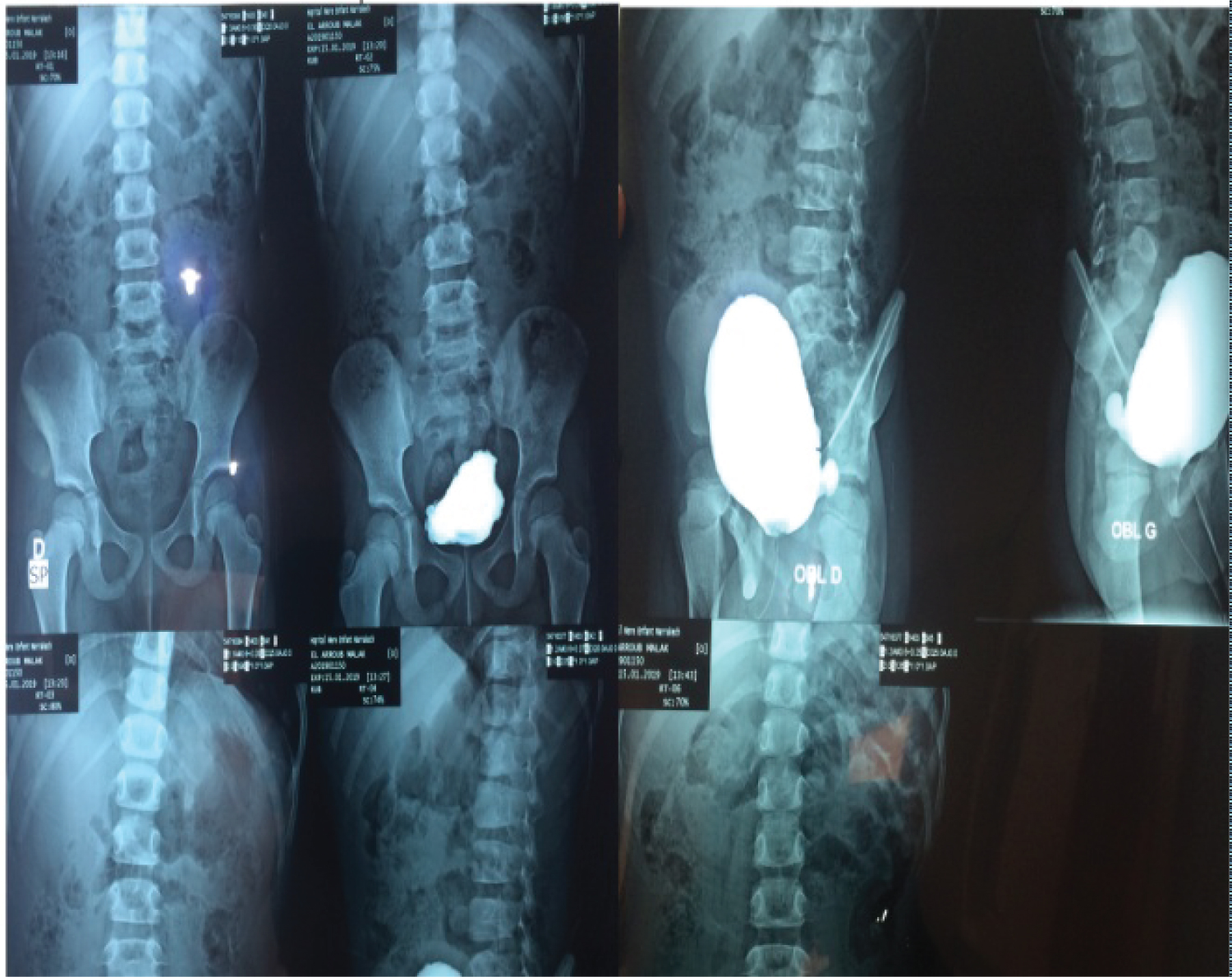 Figure 1: Urétherocystographie. View Figure 1
Figure 1: Urétherocystographie. View Figure 1
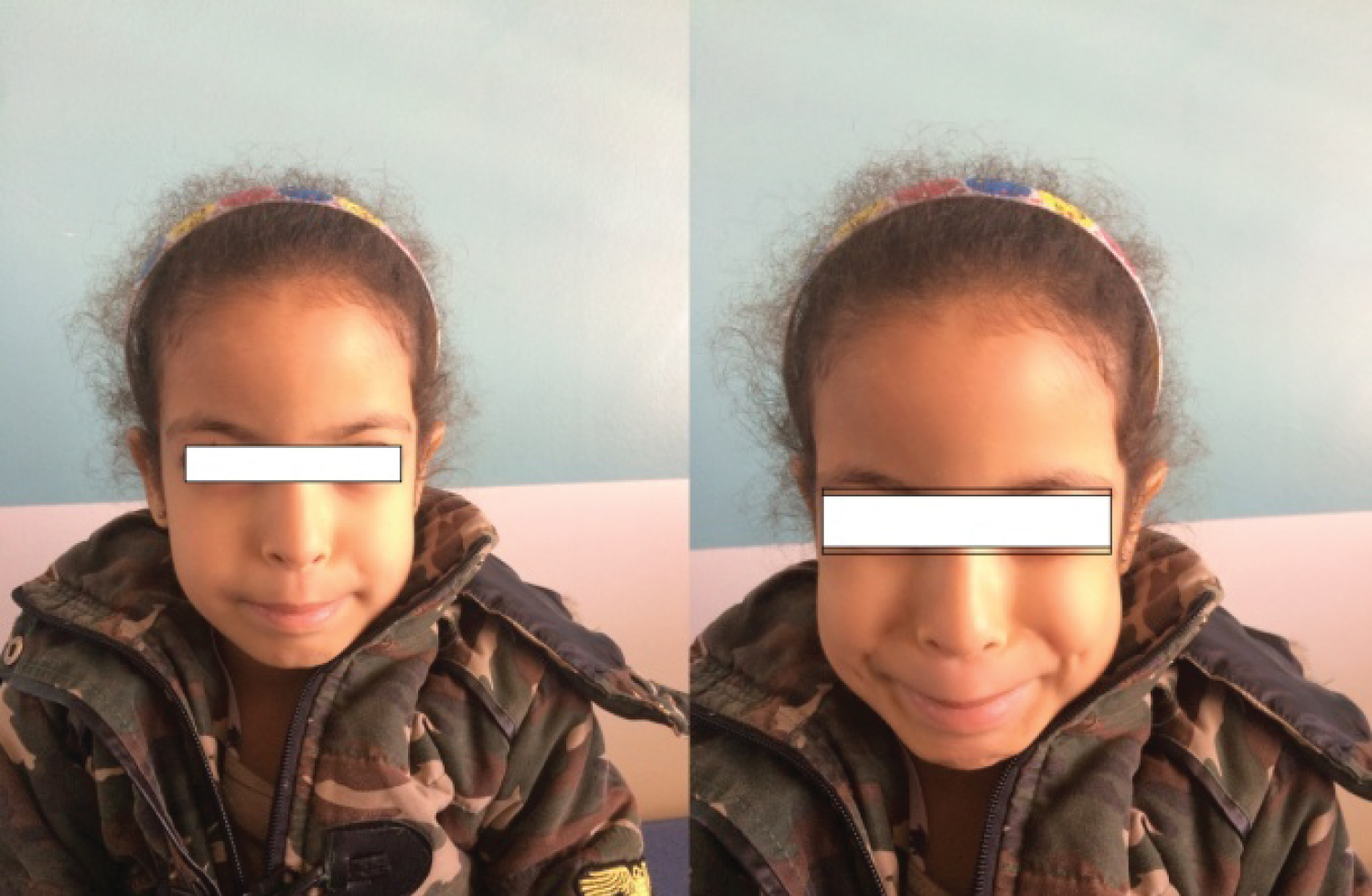 Figure 2: Facial expression of Ochoa syndrome. View Figure 2
Figure 2: Facial expression of Ochoa syndrome. View Figure 2
On the suspicion of bladder dysfunction urodynamic studies were performed. The urodynamic exploration:
Adding the urodynamic findings of a non-neurogenic bladder to the typical facial expression, the Ochoa Syndrome diagnosis was confirmed.
The genetic analysis not done due to unavailability in Morocco and high cost.
She was initially treated with antibiotic prophylaxis and anticholinergics and conservative treatment of renal failure also clean intermittent catheterization to void the bladder.
Urofacial syndrome (UFS) is characterized by prenatal or infantile onset of urinary bladder voiding dysfunction, abnormal facial movement with expression (resulting from abnormal co-contraction of the corners of the mouth and eyes), and often bowel dysfunction (constipation and/or encopresis). Bladder voiding dysfunction increases the risk for urinary incontinence, megacystis, vesicoureteric reflux, hydroureteronephrosis, urosepsis, and progressive renal impairment. In rare instances, an individual who has a molecularly confirmed diagnosis and/or an affected relative meeting clinical diagnostic criteria manifests only the characteristic facial features or only the urinary bladder voiding dysfunction (not both). Nocturnal lagophthalmos (incomplete closing of the eyes during sleep) appears to be a common and significant finding [2].
UFS was first described in several unrelated Colombian families (Elejalde, 1979; Ochoa and Gorlin, 1987). The facial abnormality is a characteristic 'inverse' facial expression when these patients smile, such that they appear to be crying. This grimacing is highly diagnostic of the UFS. Ochoa (1992) studied 50 patients from 32 families, the largest series reported so far. The majority presented with enuresis with or without urinary tract infection. A third of these patients were referred because of the characteristic facial expression, and the remainder because of urinary tract infection. Constipation was noted in over a half of the patients (28/50). Urological investigations and urodynamic studies revealed characteristic findings of neuropathic bladder in all of them: A trabeculated, large capacity bladder with narrowing of the urethral lumen in all of them; a vesicoureteral reflux in two-thirds of patients (32/50), which was bilateral in half of them (18/32); and a hypertonic, hyper reflexic type of bladder in about half of the patients (22/50). None of the parents showed the characteristic facial expression, or any abnormal finding in urological investigations.
The observation of a large number of UFS cases confined to a small geographical area could support a founder effect. However, further cases have now been reported from Kuwait (Teebi and Hassoon, 1991), and the United States (Wang, et al. 1999), as well as from Europe (Galan, et al. 1997; Chauve, et al. 2000). Furthermore, genetic homogeneity has been demonstrated in the reported North American and French families (Wang, et al. 1999; Chauve, et al. 2000). Although undoubtedly a rare condition, it seems possible that there may be cases of UFS in whom the diagnosis has not been made because of the absence of overt urinary tract symptoms [3].
UFS is a heterogeneous condition resulting from biallelic pathogenic variants in either HPSE2 or LRIG2. In some instances no pathogenic change has been identified. Note that the majority of individuals with UFS reported to date have not had molecular confirmation of their diagnosis. It is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives, prenatal testing of pregnancies at increased risk, and preimplantation genetic diagnosis are possible if the pathogenic variants in the family are known [2].
The aim of early treatment is the restoration of balanced bladder emptying and prevention of upper tract deterioration. By achieving a low-pressure and adequate bladder capacity, potential risk of upper tract dilation, associated reflux, thus, progressive renal scarring can be eliminated. Significant residual urine can cause recurrent febrile urinary tract infections. Therefore CIC is inevitable in most of the + patients. Therapeutic strategies consist of antibiotic prophylaxis, clean intermittent catheterization, anticholinergics, and intravesical botulinium toxin injection, if needed. Efficacy of the treatment varies between individuals with Ochoa syndrome (Table 1) [4].
Table 1: Urological problems, radiological findings, and management of Ochoa syndrome patients [4]. View Table 1
Monitor: For evidence of urinary tract features including vesicoureteric reflux and hydroureteronephrosis;
Renal function at intervals determined by urinary tract features at presentation and their subsequent progression;
For evidence of significant corneal involvement in individuals with nocturnal lagophthalmos.
Agents/Circumstances to avoid: Nephrotoxic substances contraindicated in individuals with renal impairment should be avoided if possible.
Evaluation of relatives at risk: It is appropriate to clarify the genetic/clinical status of sibs of an affected individual as soon as possible after birth in order to identify those who would benefit from prompt evaluation of the urinary tract and renal function and early initiation of necessary treatment.
Evaluations can include: Molecular genetic testing if the pathogenic variants in the family are known;
Examination to determine whether facial and/or urinary tract manifestations of UFS are present if the pathogenic variants in the family are not known.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy management: Although no guidelines for prenatal management of UFS exist, it seems appropriate to perform ultrasound examination of pregnancies at risk to determine if urinary tract involvement of UFS is present, as it may influence the timing and/or location of delivery (e.g., in a tertiary medical center that could manage renal/urinary complications immediately after birth) [2].
Ochoa syndrome should always be considered in patients with dysfunctional bladder who have characteristic grimacing when smiling. The prognosis of urofacial syndrome is generally poor and requires multiple treatment modalities. Early diagnosis of the syndrome is mandatory.
None.