

International Journal of Respiratory and Pulmonary Medicine
Eicosapentaenoic Acid Decreases Histamine Receptor 1 Expression on Lung Microvascular Endothelial Cells and Cell Permeability during LPS Stimulation
Takaaki Osako1, Michiko Aoyama-Ishikawa1,2, Hayato Yamashita2, Makoto Usami2, Atsunori Nakao1* and Joji Kotani1
1Department of Emergency, Disaster and Critical Care Medicine, Hyogo College of Medicine, Nishinomiya, Japan
2Department of Biophysics, Kobe University Graduate School of Health Sciences, Kobe, Japan
*Corresponding author: Atsunori Nakao, Department of Emergency and Critical Care Medicine, Hyogo College of Medicine, 1-1, Mukogawa-cho, Nishinomiya, Hyogo, 663-8501, Japan, Tel: +81-798-45-6111 E-mail: atsunorinakao@aol.com
Int J Respir Pulm Med, IJRPM-1-007, (Volume 1, Issue 1), Research Article; ISSN: 2378-3516
Received: November 27, 2014 | Accepted: December 22, 2014 | Published: December 26, 2014
Citation: Osako T, Aoyama-Ishikawa M, Yamashita H, Usami M, Nakao A, et al. (2014) Eicosapentaenoic Acid Decreases Histamine Receptor 1 Expression on Lung Microvascular Endothelial Cells and Cell Permeability during LPS Stimulation. Int J Respir Pulm Med 1:007. 10.23937/2378-3516/1410007
Copyright: ©2014 Osako T, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Abstract
Introduction: During acute lung inflammation, the lung microvasculature becomes hyperpermeable, resulting in immune cell infiltration and tissue edema. N-3 polyunsaturated fatty acids (PUFAs), when used as a supplement in parenteral nutrition, can attenuate Lipopolysaccharide (LPS)-induced lung injury. In this study, we examined the effects n-3 PUFAs on lung microvascular cell permeability.
Material and methods: Human lung microvascular endothelial cells (HMVEC-L) were seeded on fibronectin-coated transwell inserts. The cells were pretreated with docosahexaenoic acid (DHA) or eicosapentaenoic acid (EPA) (n-3 PUFAs), and then treated with LPS (1 μg/mL) to simulate acute lung injury. Microvascular permeability was assessed by monitoring the movement of FITC-albumin from the upper chamber to the lower chamber. The mRNA expression of tight junction proteins and regulators of adherens junctions were assessed by real-time PCR. VE-cadherin expression and localization was assessed by immune fluorescent staining.
Results: Pretreatment with DHA and EPA prior to LPS stimulation significantly attenuated LPS-induced cell permeability. EPA decreased histamine receptor 1 (H1R) mRNA expression following LPS stimulation. Interleukin (IL)-6 mRNA expressions in response to LPS treatment was significantly reduced by both DHA and EPA pretreatment.
Conclusions: DHA and EPA attenuated LPS-induced lung microvascular endothelial cell permeability through a mechanism that may involve IL-6. EPA pretreatment may influence H1R expression.
Introduction
Sepsis is a systemic inflammatory response to infection, leading to serious complications including acute respiratory distress syndrome and multiple organ failure. The resulting pulmonary failure can lead to morbidity and mortality in the intensive care unit (ICU) [1,2]. LPS, the main component of the cell wall of gram-negative bacteria, can induce marked acute pulmonary inflammation and lead to systemic inflammatory response syndrome or sepsis [3]. LPS increases the permeability of endothelial barriers facilitating access of white blood cells to the infected tissue, but also causing lung edema [4].
Paracellular permeability, the opening and closing of junctions between endothelial cells, is regulated primarily by Adherens Junctions (AJ) and Tight Junctions (TJ) [5]. Histamine is a strong inducer of hyperpermeability through a pathway mediated by Histamine Receptor 1 (H1R) [6]. Vascular endothelial growth factor (VEGF) promotes vascular leakage, angiogenesis, and tyrosine phosphorylation of AJ proteins [5-7]. In mice, signaling through matrix metalloprotease (MMP)-1 and protease-activated receptor-1 enhances lung vasculature permeability [8,9].
In a previous report, we demonstrated that parental nutrition containing fish oil significantly reduced LPS-induced acute lung injury as determined by reduction of neutrophil accumulation and less lung inflammation in a rat model [10]. Fish oil is rich in n-3 Polyunsaturated Fatty Acids (PUFAs), in particular Docosahexaenoic Acid (DHA) and Eicosapentaenoic Acid (EPA). Several reports have suggested that n-3 PUFAs mediate endothelial and epithelial permeability through alteration of tight junctions (TJ) [11-13]. However, there was little understanding of the relationship between factors that regulate endothelial permeability and n-3 PUFAs. The purpose of this study was to examine the effects of DHA and EPA on lung microvascular endothelial cell permeability and the expression of permeability-related genes during LPS-induced inflammation. We hypothesized that the n-3 PUFAs would mitigate LPS-induced permeability in lung microvascular endothelial cells.
Material and Methods
Cell culture
Human lung microvascular endothelial cells (HMVEC-L; Lonza, Basel, Switzerland) were grown as a monolayer in 37�C in 5% CO2 in Endothelial Growth Media-2 Microvascular (EGM™-2MV Medium; Lonza) supplemented with a SingleQuots Kit (Lonza) containing human epidermal growth factor, hydrocortisone, human recombinant fibroblast growth factor-β, vascular endothelial growth factor, insulin-like growth factor, ascorbic acid, fetal bovine serum (5%), and gentamicin/amphotericin-B.
HMVEC-L cells (10,000cells/cm2) per well were seeded on fibronectin-coated membrane transwell inserts (BD BioCoat™ Fibronectin Cellware 6.5 mm diameter inserts, 3.0 μm pore size; BD) with 100 μL culture medium added to the apical chamber and 600 μL to the basolateral chamber. The transepithelial electrical resistance (TEER) of confluent polarized HMVEC-L monolayers was measured using the EVOM resistance meter and the Endohm chamber for 6 mm culture cups (World Precision Instruments, Berlin, Germany). Readings were taken every 24 hours until the TEER had risen steadily to =45 Ωcm2, then PUFAs and/or LPS was administered.
Treatment with PUFAs and LPS
EPA, DHA, ascorbic acid (vitamin C), alpha-tocopherol (vitamin E), and LPS (E.coli O55:B5) were purchased from Sigma-Aldrich (St. Louis, MO). PUFAs were diluted in 100% ethanol to a stock concentration of 400 mM and stored at -30�C. Final PUFA concentration in the culture medium was 40 mM. Vitamins C and E (final concentrations of 75 mM and 20 mM, respectively) were added simultaneously with the PUFAs and were also administered to the control group without PUFA. Monolayers of HMVEC-L cells were incubated with EPA or DHA for 24 hours, and then fresh culture media with EPA, DHA and/ or LPS (1 μg/mL) was added to the apical camber.
Paracellular permeability assay
Paracellular permeability across the endothelial monolayers was determined by monitoring the diffusion of fluorescein isothiocynate (FITC)-conjugated albumin (Sigma). The media in the apical chamber was replaced with media containing FITC-albumin (800 μg/mL) immediately (0 hours),4 hours, and 8 hours after LPS treatment. An hour later, samples were taken from the basolateral chamber and assayed with an Infinite 200 microplate reader (Tecan Group Ltd., Germany) using excitation at 485 nm and emission at 535 nm. The data were normalized as ratio of the control group.
RNA extraction, cDNA synthesis, and real-time quantitative PCR
Total RNA was extracted from the cultured HMVEC-L with TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA was reverse-transcribed to cDNA using iScript cDNA Synthesis Kits (Biorad Hercules, CA) according to the manufacturer's instructions. Amplifications were performed by quantitative real-time RT-PCR (MyiQ, Biorad) using SYBR Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan). The primer sequences are shown in Table 1. Relative gene expression was calculated using the ddCt method after normalization to the Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) level. Because LPS-induced permeability was attenuated by DHA and EPA 4 hours after LPS stimulation, mRNA expression was determined 2 hours after LPS stimulation.
![]()
Table 1: Primer sequences for real-time RT-PCR
View Table 1
Immunofluorescent staining
Cells were seeded on fibronectin-coated 24-well culture plates. After 4 hours of LPS treatment, the cells were fixed by 4% paraformaldehyde (Sigma), washed with Tris-buffered saline (TBS), permeabilized with 2% Triton-X (Sigma) and incubated with 50 mM ammonium chloride (WAKO, Osaka, Japan). Blocking was performed with 5% bovine serum albumin (BSA) in TBS for 30 minutes. The cells were incubated overnight at 4�C with rabbit anti-VE-cadherin antibody (Abcam, Cambridge, United Kingdom). The next day, the cells were washed with TBS and incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (Cell Signaling Technology, Danvers, MA) for 1 hour at room temperature. The cells were incubated with DAPI for nuclear staining, and cell images were taken with the Nikon Ti-E microscope (Tokyo, Japan).
Statistical analysis
Data are presented as the mean + standard deviation (SD) and were analyzed by Tukey-Kramer's post hoc test. A probability level of p< 0.05 was considered statistically significant.
Results
Pretreatment of DHA or EPA inhibited the LPS-increased permeability of HMVEC-L
The permeability of HMVEC-L monolayers was determined by the flux of FITC-albumin. In the absence of LPS, DHA increased the permeability of the HMVEC-L monolayers 4 hours after treatment (Figure 1A). When inflammatory conditions were stimulated by LPS treatment, DHA and EPA significantly decreased the permeability of the endothelial monolayers at 4 and 8 hours after LPS stimulation (Figure 1B).
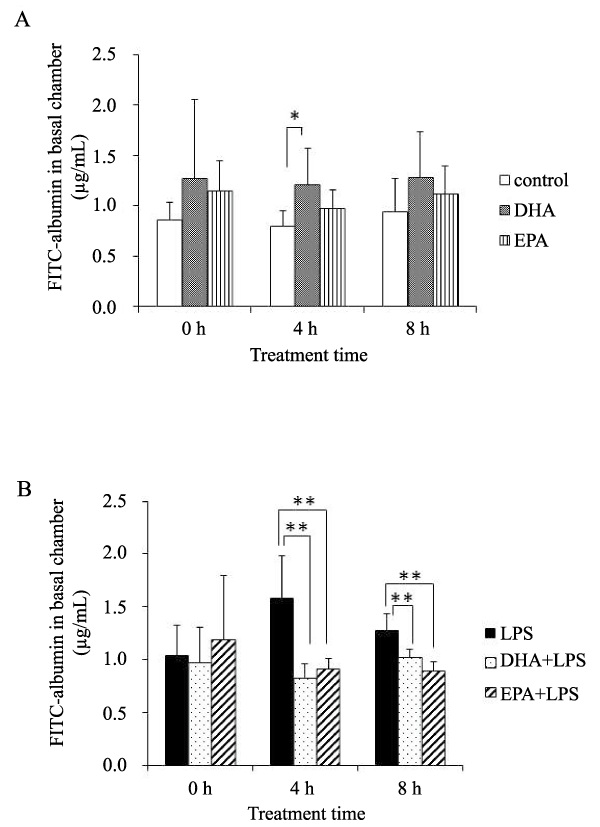
Figure 1: CTPA showing a wedge-shaped infarct in the lingula of the lung.Figure 1: Paracellular permeability as measured by the concentration of FITCalbumin in the basal chamber.
A) Sham (no LPS): White bars- control; gray bars- DHA; patterned bars- EPA, n=7 samples/group, B) LPS-treated HMVEC-L: Black bars- LPS; dotted bars- DHA+LPS; patterned bars- EPA+LPS. n=5 samples/group.
*,p< 0.05; **, p< 0.01 vs. no PUFA control by Tukey-Kramer's post hoc test.
View Figure 1
EPA decreased H1R and interleukin-6 mRNA expression in response to LPS stimulation
To investigate the mechanisms underlying modulation of lung vasculature permeability by EPA and DHA, we examined mRNA levels for several known mediators of lung vasculature permeability. In the absence of LPS treatment, EPA and DHA had no significant effects on the expression of any of the mRNAs examined (Figure 2). When LPS was administered in the absence of n-3 PUFAs, the levels of the mRNAs for H1R (Figure 2A), VEGF (Figure 2E), and interleukin (IL)-6 (Figure 2F) increased. EPA treatment significantly attenuated induction of H1R mRNA (Figure 2A), and IL-6 mRNAs in response to LPS treatment (Figure 2F). DHA significantly attenuated induction of IL-6 mRNAs in response to LPS treatment (Figure 2F).
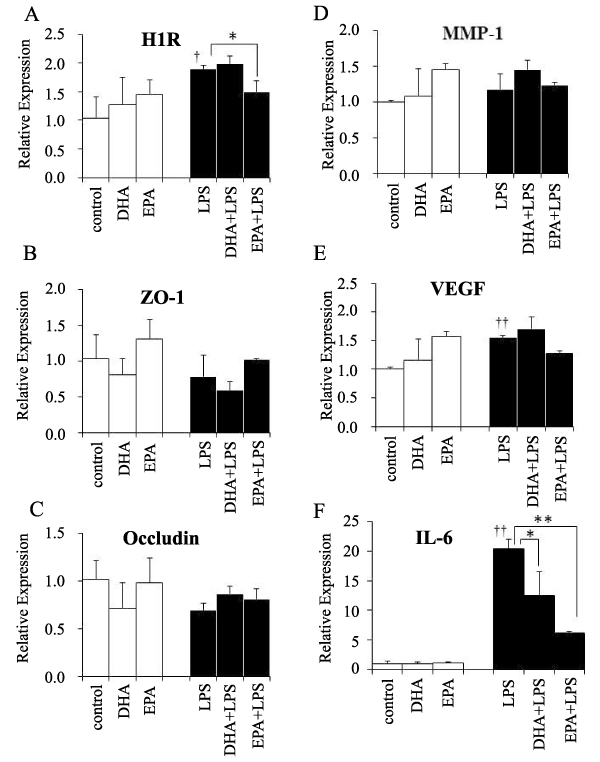
Figure 2: The mRNA levels of permeability-related genes. A) H1R mRNA, B) ZO-1 mRNA, C) occludin mRNA, D) MMP-1 mRNA, E) endothelial-derived VEGF mRNA, F) IL-6 mRNA. White bars- no LPS; black bars- LPS-treated, n=3 samples/group.
*, p< 0.05, **, p< 0.01 vs. no PUFA by Tukey-Kramer's post hoc test.
�, p< 0.05, ��, p< 0.01 vs. control by t-test.
View Figure 2
EPA attenuated LPS-decreased VE �cadherin expressions
To examine the integrity of the adherens junctions in the HMVEC-L monloayers, immunofluorescent staining was performed for VE-cadherin. In the absence of LPS treatment, VE-cadherin was localized at the intercellular gaps, and its localization was not altered by DHA or EPA treatment (Figure 3A-3C). When LPS was administered to induce inflammatory conditions, VE-cadherin was partially disrupted (Figure 3D). DHA and EPA treatment reduced the magnitude of VE-cadherin disruption in as a result of LPS treatment (Figure 3E,3 F).
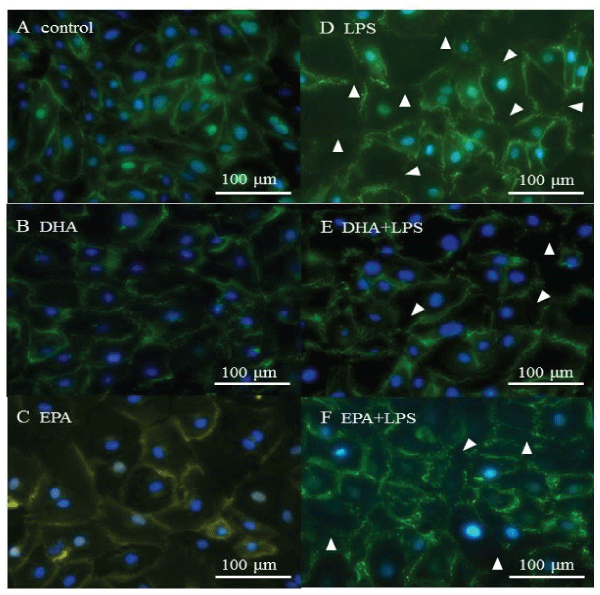
Figure 3: VE-cadherin localization. A) control, B) DHA, C) EPA, D) LPS, E) DHA+LPS, F) EPA+LPS. White arrows show disrupted VE-cadherin localization.
View Figure 3
Discussion
This study demonstrated that pretreatment with DHA or EPA attenuates LPS-induced permeability of lung microvascular endothelial cells. EPA may act through mechanisms that are H1R-dependent, while DHA likely exerts its effects through other pathways.
In the absence of inflammatory stimulation, n-3 PUFAs can influence the permeability of both epithelial and endothelial cells. DHA enhances the permeability of epithelial cells in the absence of inflammatory situation [14]. Similarly, we observed increased permeability of the HMVEC-L monolayers 4 hours after DHA treatment. It is still unclear the mechanism of increased permeability by DHA in non-inflammatory situation, however, Roig-P�rez et al. demonstrated that DHA has some influence on phospholipase C /Ca(2+)/protein kinase C pathway and it lead the disruption of epithelial barrier function in Caco-2 cells [14]. We need more study to elucidate the mechanisms of the increase of permeability by DHA.
N-3 PUFAs may prevent epithelial cell barrier disruption during inflammatory conditions by modulating TJ proteins [15]. Li and colleagues demonstrated that in T84 epithelial cells, treatment with pro-inflammatory cytokines (TNF-α and IFN-γ) decreased ZO-1 and occluding expression, and concurrent treatment with n-3 PUFAs increased expression of these TJ protein and improved cell barrier function [15]. In our study DHA and EPA attenuated LPS-induced cell permeability, similar to the results of Li and colleagues; however, neither DHA nor EPA changed ZO-1 or occludin mRNA expression. The use of endothelial cells rather than epithelial cells may explain this difference. Endothelial cells express VE-cadherin as a specific adhesion molecule, and the AJ is a key regulator of endothelial cell permeability [16]. In our study, DHA and EPA attenuated LPS-induced disruption of VE-cadherin, and EPA significantly attenuated H1R mRNA upregulation during LPS stimulation. Histamine can induce a transient and reversible disruption of AJ proteins, such as β-catenin and VE-cadherin, leading endothelial cell hyperpermeability [17]. Thus, in lung endothelial cells, DHA and EPA may influence AJ function more strongly than TJ function.
The anti-inflammatory properties of n-3 PUFAs are well-established, and n-3 PUFAs regulate nuclear factor ?-B, an important regulator of IL-6 expression [18]. In the HMVEC-L, we detected anti-inflammatory effects of DHA and EPA as decreased expression of IL-6 mRNA following LPS treatment. Because IL-6 enhances endothelial permeability [19], reduction of IL-6 levels may also explain why DHA and EPA reduced the HMVEC-L permeability. In addition, there are several reports that histamine induceIL-6 expression [20]. Thus, the reduction of H1R mRNAs following EPA treatment may influence IL-6 expression.
In summary, we demonstrated that DHA and EPA can attenuate LPS-induced permeability of lung microvascular endothelial cells. We hypothesize that reduction of IL-6 expression may be an underlying mechanism. Reduction of H1R F level by EPA may also have important roles and may alter the expression of VE-cadherin and IL-6.This is the first report suggesting that n-3 PUFAs may have effects on H1R during inflammation that affects lung microvascular cell permeability. We are continuing to study the underlying mechanism to better understand how n-3 PUFA-based treatments, particularly with EPA, may reduce sepsis-mediated pulmonary injury in ICU patients.
Acknowledgment
This research was supported by JSPS KAKENHI Grant Number 24791956.
References
-
Ware LB, Matthay MA (2000) The acute respiratory distress syndrome. N Engl J Med 342: 1334-1349.
-
Eisner MD, Thompson T, Hudson LD, Luce JM, Hayden D, et al. (2001) Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J RespirCrit Care Med 164: 231-236.
-
Altemeier WA, Matute-Bello G, Gharib SA, Glenny RW, Martin TR, et al. (2005) Modulation of lipopolysaccharide-induced gene transcription and promotion of lung injury by mechanical ventilation. J Immunol 175: 3369-3376.
-
Shapiro H, Kagan I, Shalita-Chesner M, Singer J, Singer P (2010) Inhaled aerosolized insulin: a "topical" anti-inflammatory treatment for acute lung injury and respiratory distress syndrome? Inflammation 33: 315-319.
-
Goddard LM, Iruela-Arispe ML (2013) Cellular and molecular regulation of vascular permeability. ThrombHaemost 109: 407-415.
-
Niimi N, Noso N, Yamamoto S (1992) The effect of histamine on cultured endothelial cells. A study of the mechanism of increased vascular permeability.Eur J Pharmacol 221: 325-331.
-
Harris ES, Nelson WJ (2010) VE-cadherin: at the front, center, and sides of endothelial cell organization and function. CurrOpin Cell Biol 22: 651-658.
-
Shen L, Black, ED, Witkowski ED,Lencer WI, Guerriero V, Schneeberger EE, Turner JR (2006) Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J. Cell Sci119: 20952-106.
-
Tressel SL, Kaneider NC, Kasuda S, Foley C, Koukos G, et al. (2011) A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol Med 3: 370-384.
-
Kohama K, Nakao A, Terashima M, Aoyama-Ishikawa M, Shimizu T, et al. (2014) Supplementation of parenteral nutrition with fish oil attenuates acute lung injury in a rat model. J ClinBiochemNutr 54: 116-121.
-
Li Q, Zhang Q, Zhang M, Wang C, Zhu Z, et al. (2008) Effect of n-3 polyunsaturated fatty acids on membrane microdomain localization of tight junction proteins in experimental colitis. FEBS J 275: 411-420.
-
Beguin P, Errachid A, Larondelle Y, Schneider YJ (2013) Effect of polyunsaturated fatty acids on tight junctions in a model of the human intestinal epithelium under normal and inflammatory conditions. Food Funct4: 923-931.
-
Oh-I S, Shimizu H, Sato T, Uehara Y, Okada S, Mori M (2005) Molecular mechanisms associated with leptin resistance: n-3 polyunsaturated fatty acids induce alterations in the tight junction of the brain. Cell Metab 1: 331-341.
-
Roig-P�rez S, Cortadellas N, Moret� M, Ferrer R (2010) Intracellular mechanisms involved in docosahexaenoic acid-induced increases in tight junction permeability in Caco-2 cell monolayers. J Nutr 140: 1557-1563.
-
Li Q, Zhang Q, Wang M, Zhao S, Xu G, et al. (2008) n-3 polyunsaturated fatty acids prevent disruption of epithelial barrier function induced by proinflammatory cytokines. MolImmunol 45: 1356-1365.
-
Crosby CV, Fleming PA, Argraves WS, Corada M, Zanetta L, et al. (2005) VE-cadherin is not required for the formation of nascent blood vessels but acts to prevent their disassembly. Blood 105: 2771-2776.
-
Guo M, Breslin JW, Wu MH, Gottardi CJ, Yuan SY (2008) VE-cadherin and beta-catenin binding dynamics during histamine-induced endothelial hyperpermeability. Am J Physiol Cell Physiol 294: C977-984.
-
Singer P, Shapiro H, Theilla M, Anbar R, Singer J, Cohen J (2008) Anti-inflammatory properties of omega-3 fatty acids in critical illness: novel mechanisms and an integrative perspective. Intensive Care Med 34:1580-1592.
-
Desai TR, Leeper NJ, Hynes KL, Gewertz BL (2002) Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res 104: 118-123.
-
Li Y, Chi L, Stechschulte DJ, Dileepan KN (2001) Histamine-induced production of interleukin-6 and interleukin-8 by human coronary artery endothelial cells is enhanced by endotoxin and tumor necrosis factor-alpha Microvasc Res 61: 253-262.
