The complexity of the musculoskeletal and rheumatologic manifestations of hematologic disease, both benign and malignancy-associated haematologic disease, is extensively explored in this accompanying review article. Accompanying reference to unique radiological images in this review aids in understanding the importance and effect of hematological disease on patient quality of life. This review implores the understanding of the “immune-skeletal interface” as the unique basis of osteoimmunology, as well as descriptively accounting for avenues of bone health to be observed when treating the patient with hematologic disease.
ISI: Immune-Skeletal Interface, Osteoimmunology, Bone health, Hematological disease
AA: Aplastic Anaemia; AHA: Acquired Haemophilia A; ALAS2I : delta-Aminolevulinate Synthase 2; ALL: Acute Lymphoblastic Leukemia; AL: Systemic Light Chain Amyloidosis; AML: Acute Myeloid Leukemia; ASCT: Allogeneic Stem Cell Transplant; ATG: Anti-Thymocyte Globulin; ATLL: Adult T-Cell Leukemia-Lymphoma; AV: Arteriovenous; AVN: Avascular Necrosis; CAMT: Congenital Amegakaryocytic Thrombocytopenia; CD: Cluster Of Differentiation; CIT: Chemoimmunotherapy; Chl: Classic Hodgkin Lymphoma; DNA: Deoxyribonucleic Acid; DBA: Diamond Blackfan Anaemia; DGS: Digeorge Syndrome; DKC: Dyskeratosis Congenita; DLBCL: Diffuse Large B-Cell Lymphoma; EN: Extra-Nodal; EM: Extra-Medullary; FA: Fanconi Anaemia; FANC: Fanconi Anemia Complementation Group; FDG PET/CT: F- 18 Fluorodeoxyglucose Positron Emission Tomography/Computed Tomography; GVHD: Chronic Graft-Versus-Host Disease; HL: Hodgkin Lymphoma; HIV: Human Immunodeficiency Virus; HIES: Hyperimmunoglobulin E Syndrome; Ige: Immunoglobulin E; ISI: Immune-Skeletal Interface; IBMFS: Inherited Bone Marrow Failure Syndromes; IGF-1: Insulin-Like Growth Factor-1 Axis; IST: Immune Suppressive Therapy; ITP: Immune Thrombocytopaenia; MDS: Myelodysplastic Syndrome; MCPJ: Metacarpophalangeal Joints; MMP: Myeloid Malignancy Predisposition Syndromes; MRI: Magnetic Resonance Imaging; NTD: Neural Tube Defects; NGS: Next Generation Sequencing; NLPHL: Nodular Lymphocyte Predominant Hodgkin Lymphoma; NHL: Non-Hodgkin Lymphoma; OPG: Osteoprotegerin; RA: Rheumatoid Arthritis; RP: Ribosomal Protein; PTT: Partial Thromboplastin Time; PCT: Porphyria Cutanea Tarda; PMF: Primary Myelofibrosis; PCV: Polycythemia Vera; PET: Positron Emission Tomography; PBL: Plasmablastic Lymphoma; PIP: Proximal Interphalangeal; PSS: Primary Sjogren Syndrome; SLE: Systemic Lupus Erythematosus; SDS: Shwachman-Diamond Syndrome; SCN: Severe Congenital Neutropenia; SRE: Skeletal Related Events; TFR: Treatment Free Remission; TAR: Thrombocytopenia Absent Radii; TAO: Thalassemia-Associated Osteoporosis; TNF: Tumour Necrosis Factor; TKIS: Tyrosine Kinase Inhibitors; RANKL: Receptor Activator For Nuclear Factor Kappa B Ligand; VCFS: Velocardiofacial Syndrome; WHO: World Health Organization
Haematological involvement of the musculoskeletal system is not limited to the bone and joints but involves the central and peripheral nervous system, either directly or indirectly. Involvement of the central and peripheral nervous system may be in the form of mass effect in the case of compressive lesions of the spinal cord and the peripheral nerves, pathological fractures of the vertebra with encroachment on the spinal cord as well as primary intracerebral mass lesions in lymphomas of immune privileged sites. Peripheral neuropathies with distal weakness and impaired mobility are also not uncommon in haematological disease with associated disease processes referred to later in this review.
Haematolgical disease affecting the musculoskeletal system is either inherited with characteristic somatic aberrations noted from birth or is acquired through complex clonal processes resulting in the development of lymphomas. The sequelae of various treatment modalities applied in haematologic disease whether pharmacologic (chemoimmunotherapy) or non-pharmacological (radiation therapy) also have untoward effects on overall bone metabolism and bone health with some of these disease processes referred to in this review [1].
Alterations in bone mineral density represents the dissociation in the relationship between new bone formation (osteogenesis) and bone breakdown (osteoclastogenesis) with alterations in this homeostasis incurred in haematological disease. Alterations in homeostasis favouring osteoclastogenesis with accompanying reduced bone mineral density predisposes chronically ill haematology patients to insufficiency fractures [1]. Compounding variables implicated in increased bone turnover include traditional risk factors implicated in osteoporosis, those being ageing, post-menopausal states, chronic use of ethanol and tobacco as well as co-morbid HIV infection 1.
An important molecular interaction of haematopoietic cells and elements of the skeletal system occurs at the immune-skeletal interface (ISI). Other bodies of work have coined the study of this interface as “osteoimmunology” and have highlighted the evolutionary synchronous co-development of the acquired immune system and skeletal system as an explanation as to the close relationship between bone and the immune system 2. The immune-skeletal interface (ISI) highlights the importance of the dual role that immune cells play in the regulation of inflammation and the process of bone remodeling. This complex linkage between haematopoietic cells and bone health is established through several different pathogenic mechanisms including 1) Growth factors (macrophage colony stimulating factor/M-CSF) and their effect on osteoclast activation, 2) The role of B-lymphocytes in the production of osteoprotegerin (OPG) and OPG’s role in moderating receptor activator for nuclear factor kappa B ligand (RANKL) activity and 3) The ability of activated T-lymphocytes to mediate osteoclastogenesis via RANKL and tumour necrosis factor (TNF) [1,2]. Mature osteoblasts also produce IL-17 supporting lymphocyte progenitors with Notch ligand delta-like 4 production by osteoblasts regulating T-cell progenitors 2. The relationship between the bone milieu and lymphocyte function is further demonstrated by bone loss and fracture risk which accompanies altered T-helper cell function in patients with human immunodeficiency virus (HIV) as well as the role of cytokines in accelerated bone turnover that occurs in rheumatoid arthritis (RA) 1. Figure 1 below lists haematologic diseases with osteolytic, osteoblastic and osteosclerotic bone lesions.
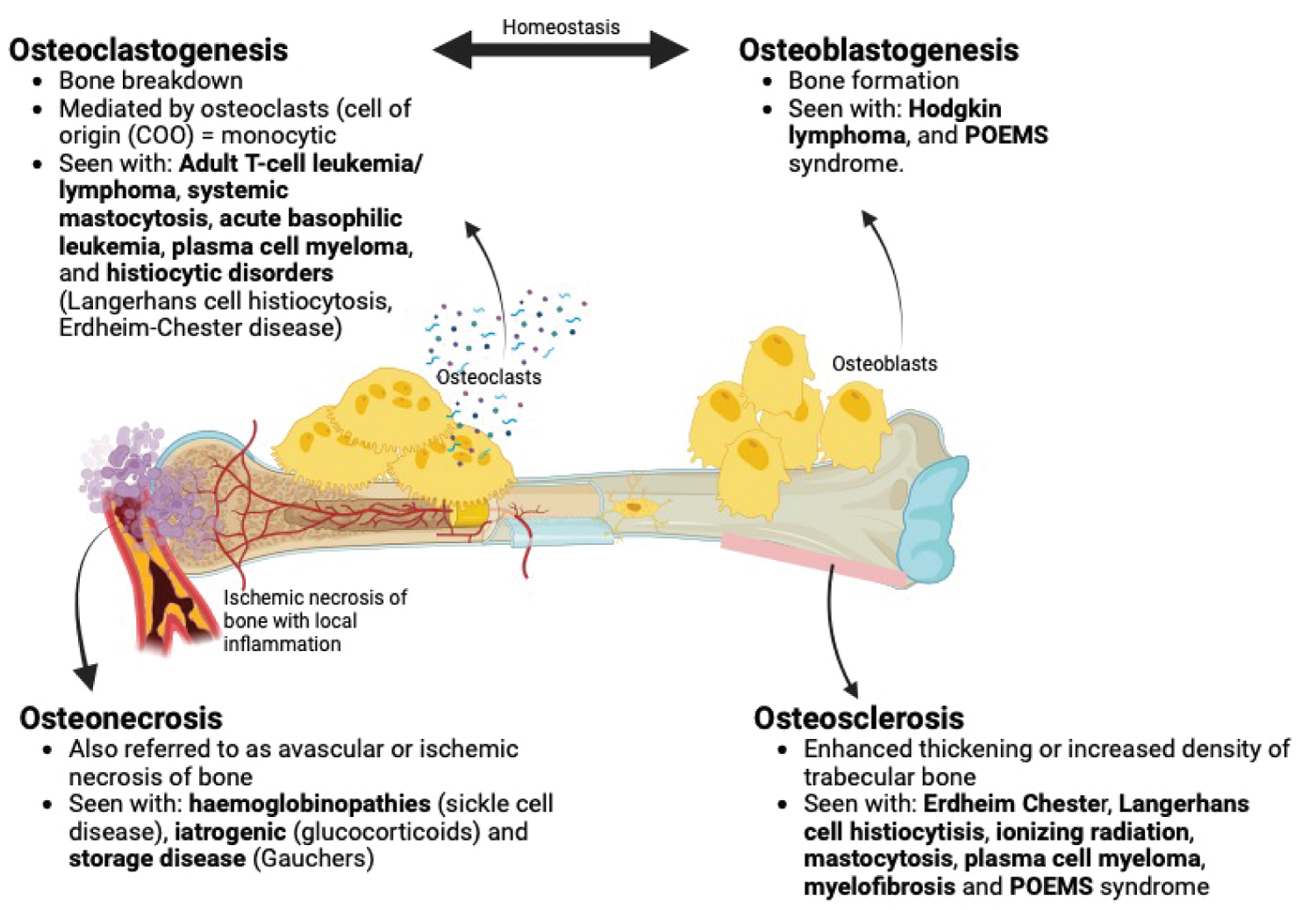 Figure 1: Haematological disease and their patterns of bone involvement 3 (image created using BioRender).
View Figure 1
Figure 1: Haematological disease and their patterns of bone involvement 3 (image created using BioRender).
View Figure 1
Our understanding of the key elements involved in the immune-skeletal interface will help to understand the role that lymphocytes play in the regulation of bone metabolism in both health and disease states. The mechanisms by which systemic risk factors (age, smoking history, alcohol use) work in concert with disease states may also serve as a target for potential therapeutic manipulation in the prevention of bone mineral density loss 1. Exploring the role and the extent to which monoclonal antibodies alter B-cell lymphocyte populations and bone metabolism in B-cell lymphomas is also a potentially unexplored aspect in the complexity of the ISI 1.
While this review does not explore deeper molecular aberrations incited by each of the disease entities discussed below, it does implore a greater consideration for the interaction between haematological disease states and the musculoskeletal system.
Inherited one marrow failure syndromes (IBMFS) and the myeloid malignancy predisposition syndromes (MMP)
Immunodeficiency syndromes
Haemoglobinopathies
Cold autoimmune haemolytic anaemias and cryoglobulinemia
Iron overload
The Porphyrias
The use of the word “benign” in this review implies a non-neoplastic process but does not imply that the condition or disease is without significant morbidity. Conditions labelled “benign”, as in the inherited bone marrow failure syndromes (IBMFS), have the potential to progress to neoplastic disorders, including haemato-lymphoid or solid organ malignancies.
The inherited bone marrow failure syndromes are: Fanconi Anaemia (FA), Diamond Blackfan Anaemia (DBA), Shwachman-Diamond (SDS), Dyskeratosis congenita (DKC), severe congenital neutropenia (SCN), congenital amegakaryocytic thrombocytopenia (CAMT) and thrombocytopenia absent radii syndrome (TAR).
The inherited bone marrow failure syndromes come to attention predominantly in childhood as they present with certain characteristic musculoskeletal anomalies accompanied by cytopenias in one or more of the cell lines.
Characterizing the IBMFS according to molecular aberrations reveals four major sub-groupings: 1) Abnormalities in DNA damage repair response in the case of FA; 2) Defects in ribosome function or “ribosomopathies” (DBA and SDS;), 3) Telomere disease (“telomeropathies”) with altered telomere function (DKC) and 4) Alterations in function of haemopoietic growth factors. These syndromes may also be classified according to the resultant cytopenia incurred namely pancytopaenia (FA, DKC), anaemia (DBA), neutropenia (SCN, SDS) and thrombocytopenia (CAMT, TAR) [3,4].
Fanconi anaemia, traditionally suspected on screening by demonstrating chromosomal breakage in the presence of DNA cross-linking agents, is best diagnosed with molecular testing with next generation sequencing (NGS) platforms [5,6]. The predominant inheritance pattern of FA is autosomal recessive or, rarely X-linked recessive (Fanconi anaemia complementation group gene B or FANCB ) , with higher incidences described in patients of Ashkenazi Jewish descent and those of Afrikaner ancestry [5-7]. The birth incidence of FA in black South Africans approximates 1 in 40 000. Mutations in FANCA, FANCC and FANCG genes account for greater than 90% of cases, with abnormalities in South African populations occurring in FANCG gene in black patients and FANCA in individuals of Afrikaner ancestry [7,8].
The clinical triad of anomalies described in FA are somatic, haematological and oncological with significant disease morbidity and mortality related to the development of myelodysplastic syndrome (MDS), aplastic anaemia (AA) and clonal evolution to leukaemia (AML) 8. FA is also regarded as a predisposing factor to solid malignancies of the head and neck, liver, oesophagus and female genital tract 8.
Data collected over a period of 43 years from 1974 to 2017 at Universitas Hospital in Bloemfontein (Free State Province, South Africa) of 144 confirmed cases of FA, noted the following noteworthy somatic abnormalities: 1) Greater than 70% of the study population were below the 10 th percentile for weight, height and head circumference, 2) 81.9% patients had documented abnormal thumbs or hands, and 3) Characteristic facies (small eyes, low hairline, triangular face and puckered mouth) were observed in 84.6% of patients 6.
Occasionally the bone marrow findings in DKC: Dyskeratosis Congenita precede the described mucocutaneous features. Osteoporosis, aseptic necrosis and scoliosis are among the described skeletal anomalies which occur in around 5% of patients with DKC with a further 19% of patients with short stature [9]. A case report from Morrison, documented physical findings in the first two recognized cases of DKC in South Africa, with buccal mucosal changes, “reticulated” skin pigmentation and dystrophic nail changes among some of the physical changes reported [10].
Diamond Blackfan Anaemia , characterized by erythroblastopenia, is driven by mutations in one of several ribosomal protein (RP) genes [11]. Whilst the incidence of DBA in South Africa is unknown, data from international registries report an incidence of between 1:100,000 to 1:200,000 live births. With approximately 1,000,000 live births reported annually in South Africa one could expect that between five and ten children with DBA would be born annually. A feature of over half of those affected by DBA include a number of skeletal, cardiac and urogenital anomalies. Skeletal malformations are divided into those more cranial/cephalad (cleft palate) and those of the extremities, in particular the thumbs. The described thumb anomalies are the rarely reported but classic triphalangeal thumb with other aberrations described including bifid thumbs and hypoplastic thumbs. Growth retardation, short stature and syndactyly are also reported in DBA [12].
Congenital amegakaryocytic thrombocytopaenia (CAMT) may present with skeletal anomalies in the form of limited pronation/supination of the forearm and hip dysplasias. Central nervous system (CNS) abnormalities reported in CAMT include cerebral and cerebellar hypoplasia [9].
Those affected with Thrombocytopenia absent radii (TAR) syndrome present with defects resembling FA, albeit less frequently documented compared to FA. As the name states, the radii are absent with intact yet malformed thumbs 13. Other reported anomalies are short or absent ulnae, absent humeri, dislocated hips and abnormal knees 13.
The myeloid malignancy predisposition syndromes (MMP) , SAMD9/SAMD9L disorder and its manifestations are remembered by the mnemonic MIRAGE. MIRAGE encompasses myelodysplasia, infection, restriction of growth, adrenal hypoplasia, genital anomalies and enteropathy. This disease entity is due to a gain of function mutation in SAMD9 leading to a multisystemic growth restriction disorder [14].
Hyperimmunoglobulin E syndrome (HIES) is characterized by a number of immunological aberrations and their consequences including defective neutrophil chemotaxis with mild neutropenia, defective T-helper lymphocyte function, recurrent sinopulmonary and skin infections (Staphylococcus aureus, Candida) and raised serum immunoglobulin E levels. Classically described non-immune findings are the characteristic facies (facial asymmetry, broad nose, deep-set eyes), retained primary teeth, recurrent fractures induced by minimal trauma (long bones, ribs), scoliosis, osteopenia and limb-length discrepancies [15].
Schimke immune-osseous dysplasia, cartilage hair hypoplasia and phosphoglucomutase 3 deficiency are other notable immunodeficiencies with skeletal aberrations [16].
Sickle cell disease (SCD) produces skeletal events mediated by a complex interplay between altered red cell rheology and altered endothelial function in a number of vascular beds. In young children, vaso-occlusion with subsequent infarction of the small bones of the hands and feet produces swelling in the dorsal aspects of these areas resulting in dactylitis or “hand-foot” syndrome [17]. Swelling in these areas is mediated by extensive infarction of the marrow, the medullary trabeculae and inner layer of cortical bone together with new bone formation in the sub-periosteal region. Episodes of dactylitis that occur before the age of five are a strong predictor of more severe disease later in life [18].
Vaso-occlusion with resultant bone infarction also predisposes the patient with sickle cell disease to the development of osteomyelitis. Micro-organisms implicated in osteomyelitis are Salmonella species and Staphylococcus aureus with reported infection with the former organism being twice as common [17,18]. In the acute setting, symptomatology of osteomyelitis may mimic that of acute bony infarctions and is an important differential to exclude [18].
The femoral and humeral joints are particularly vulnerable to ischemic necrosis (avascular necrosis) in sickle cell disease with destruction of the articular surfaces of the femoral heads producing chronic intractable pain and functional limitation [19].
The spinal radiograph (Figure 2) shows vertebral bodies displaying the “fish-vertebra” sign. In SCD these are also called H-shaped vertebra and are due to central end plate depression [20]. This sign is described in SCD when the exaggeration of the normal concavity of the superior and inferior surfaces of one or more vertebral bodies is noted on X-ray. Vertebral concavities are ascribed to micro-infarcts of the central portions of the vertebral growth plates [21].
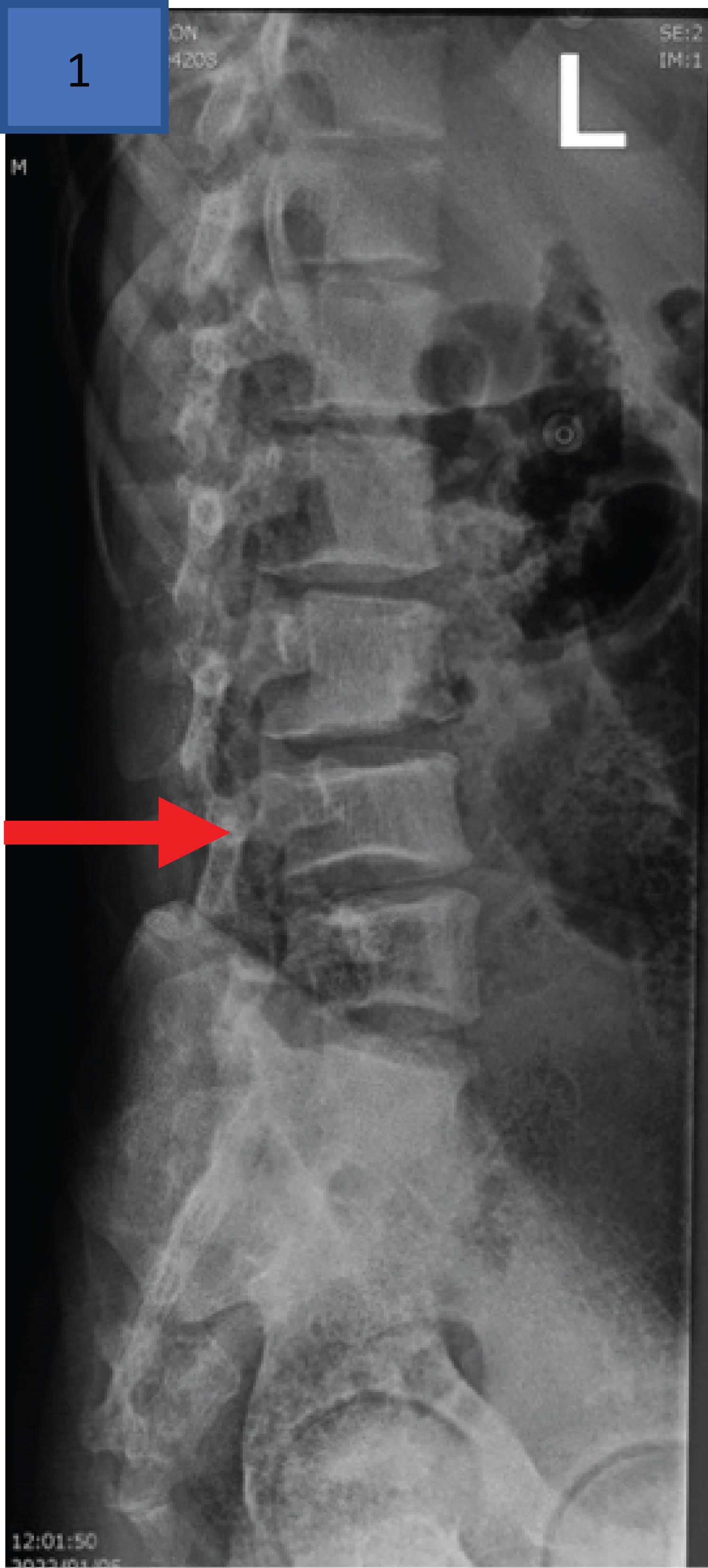 Figure 2: Lateral radiograph of the lumbar spine of a 16-year-old male with homozygous SCD with H-shaped L4 vertebra secondary to central end plate depressions (red arrow indicates this “fish-vertebra” sign) 21.
View Figure 2
Figure 2: Lateral radiograph of the lumbar spine of a 16-year-old male with homozygous SCD with H-shaped L4 vertebra secondary to central end plate depressions (red arrow indicates this “fish-vertebra” sign) 21.
View Figure 2
Beta thalassemias, typified by reduced production of β-globin chains and alterations in haemoglobin production, drive the central tenant of the disease, namely ineffective erythropoiesis. Ineffective erythropoiesis ultimately drives expansion of haemopoietic tissue in marrow expanses resulting in the typical skeletal manifestations of this disease including craniofacial protrusions and/or haemopoietic pseudotumor formation. Radiography of the skull in patients without adequate chronic transfusion support may produce a “hair-on-end” appearance due to bone marrow expansion through the thin cortical bone of the skull [22]. Expansion of haemopoietic tissue within the marrow results in pain or in cases of pseudotumor formation compressive symptomatology dependent on site of involvement [23]. Other conditions associated with ineffective erythropoiesis and extramedullary haemopoiesis of the facial bones include sideroblastic anaemia, X-linked sideroblastic anaemia and delta-aminolevulinate synthase 2 ( ALAS2) mutations [22].
Patients with thalassemia, in particular children and adolescents, who remain untreated or receive inadequate transfusion therapy are particularly vulnerable to restricted growth and under-developed musculature [24].
Osteoporosis in thalassaemic patients (thalassemia-associated osteoporosis (TAO)) occurs in patients despite appropriate therapy for their disease [25]. The life-time fracture risk, predicted at 71% in this cohort of patients, is driven by an interplay between the chronic anaemia, primary iron overload, associated endocrinopathies from iron injury to the pituitary, accompanied with an aging population of thalassemia patients [22,23,25]. The severity of the thalassemia syndrome, based on transfusion dependence seems to be a fracture risk [25].
Low bone mass and osteoporosis increase the risk of insufficiency fractures and occur in up to 90% of patients affected by thalassemia with more frequent occurrence in non-transfusion dependent thalassemia (NTDT). Contributors to decreased bone mineral density in thalassemia include iron overload with osteoblast toxicity, ineffective erythropoiesis, hypogonadism, iron chelation with desferrioxamine (DFO), vitamin D deficiency, hypercalciuria and a reduction in weight-bearing exercises. Hypogonadotropic hypogonadism (secondary hypogonadism) occurs in over half of patients with thalassemia and in children this manifests as delayed puberty. Secondary hypogonadism in adults results in decreased libido, infertility, and osteoporosis [25].
Chronic leg ulceration is not unique to haemolysis in sickle cell disease (SCD) but has its associations with other haemolytic anaemias (thalassemia, hereditary spherocytosis) with coincidental associations described with hereditary elliptocytosis and pyruvate kinase deficiency [26]. Risk factors predisposing to ulcer formation in SCD as well as the pathogenesis of delayed healing in these ulcers are alluded to in Figure 3 [27]. Venographic studies in patients with SCD have found that venous insufficiency is not the primary driver of lower limb ulceration instead it is arteriovenous (AV) shunting and reduced oxygenation of overlying skin that promotes tissue devitalization [27].
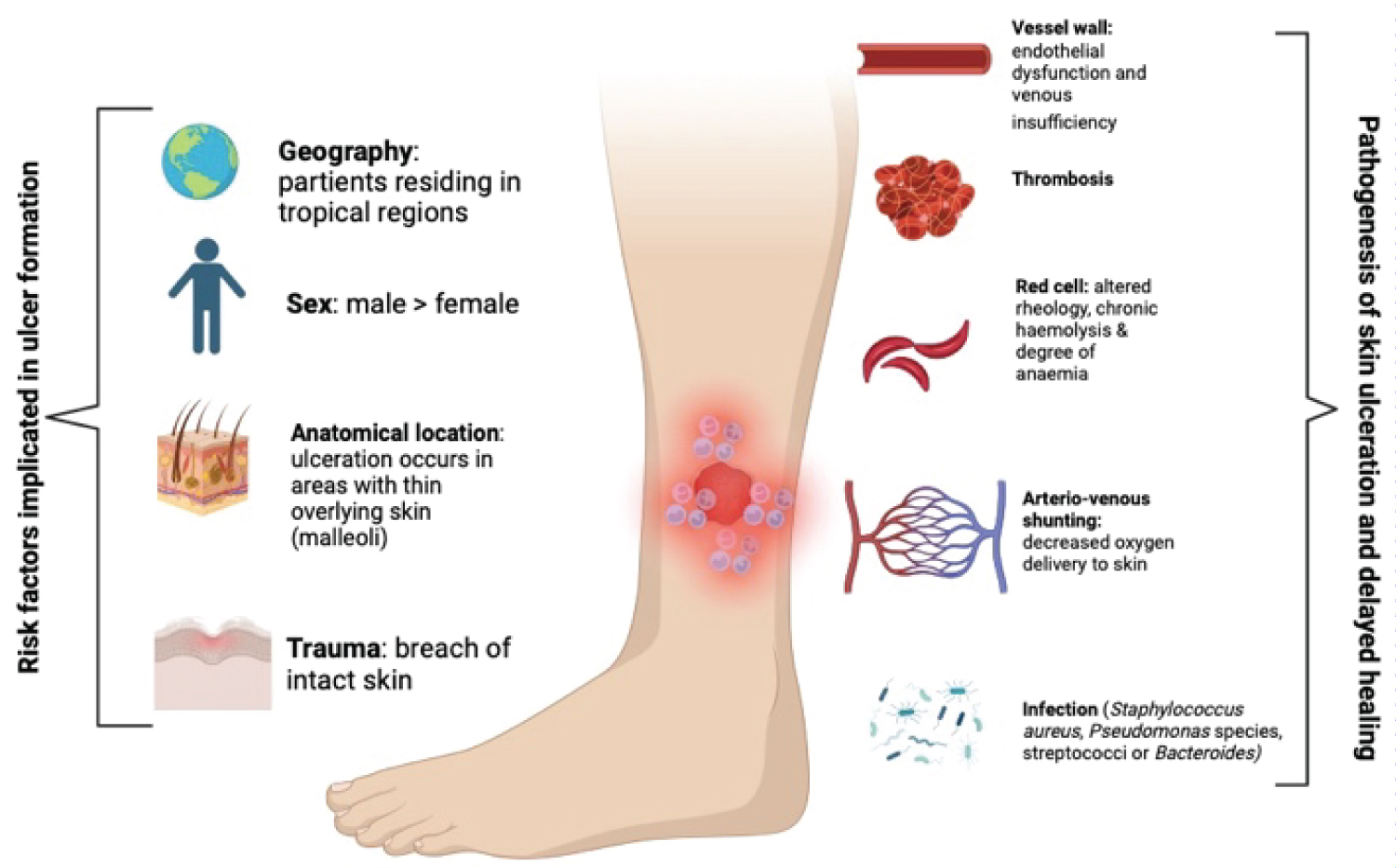 Figure 3: Leg ulcers in sickle cell disease - risk factors implicated in ulcer formation as well as pathogenesis of delayed healing [19] (image adapted from Vichinsky [19] and created using BioRender.com).
View Figure 3
Figure 3: Leg ulcers in sickle cell disease - risk factors implicated in ulcer formation as well as pathogenesis of delayed healing [19] (image adapted from Vichinsky [19] and created using BioRender.com).
View Figure 3
Three quarters of males affected with Alpha-thalassemia X-linked intellectual disability (ATR-X) syndrome have evidence of alpha thalassemia with red cell indices either in the normal range or with a microcytic hypochromic blood picture. Haemoglobin H inclusions in this disease are demonstrated on supravital stains. Distinctive cranio-facial abnormalities accompany this syndrome which include a small head circumference, hypertelorism, short triangular nose, tented upper lip with coarsening of facial features seen over time [28,29].
Cold agglutinin disease (CAD), a haemolytic anaemia driven primarily by IgM Kappa antibodies produced by clonal B cells, produces ischemic symptoms within the extremities. These ischemic symptoms manifest either as Raynauds phenomenon or acrocyanosis [30].
A similar process to cold agglutinin disease in its predilection for involving the extremities however dissimilar in its lack of haemolysis, is cryoglobulinemia . In type one cryoglobulinemia, monoclonal immunoglobulins (IgG or IgM) are produced in the following conditions: Monoclonal gammopathy of undetermined significance (MGUS), Waldenstroms macroglobulinemia, multiple myeloma and chronic lymphocytic leukemia [31]. Extremity and skin changes are reportedly common in type one cryoglobulinemia and include palpable purpura, livedo reticularis, Raynaud's phenomenon, acrocyanosis, skin necrosis, ulcers and infarcts of the digits 31. Patients with this disease process suffer with painful peripheral neuropathies confined to the extremities with sensory impairment predominating 31.
Manifestations of iron overload whether due to inherent disease ( haemochromatosis ) or secondary to chronic transfusion therapy ( siderosis ) is marred by chronic iron overload with excess of iron unused by the erythron deposited in endocrine glands, cardiac tissue, and the liver. Up to 80% of patients with haemachromatosis develop a chronic progressive non-inflammatory arthropathy typically affecting the 2 nd and 3 rd metacarpophalangeal joints (MCPJ) with proximal interphalangeal joints (PIP), knees, wrists, hips, shoulders and ankles also affected. The arthropathy of haemochromatosis is complex with joint pain and stiffness rarely resolved with chelation [32].
Porphyria cutanea tarda (PCT) is one of the more prevalent of the porphyrias and is due to inhibition of uroporphyrinogen decarboxylase, a key enzyme in heme biosynthesis. Disease onset occurs in the 5 th decade of life with skin fragility and chronic blistering skin lesions reported [33]. Scleroderma-like skin changes with characteristic dystrophic calcifications are uncommon and tend to occur in the chronic stages of the disease. Skin biopsy is unfortunately unable to morphologically distinguish PCT from systemic sclerosis (SSc) or morphea thus making the measurement of porphyrin levels essential for diagnosis [34]. In cases of congenital erythropoietic porphyria skin thickening, areas of skin hypo and hyper-pigmentation with hypertrichosis of the face and extremities is seen. Areas of skin are predisposed to secondary infection with focal bone resorption resulting in disfigurement of bony areas of the face and hands [35].
Thrombocytopenia with macrothrombocytopenia
The haemophilias
Acquired Haemophilia
DiGeorge syndrome (DGS/22q11.2 deletion) also known as velocardiofacial syndrome (VCFS) with an estimated prevalence of between 1:2000-1:6000 live births, has a range of phenotypic anomalies. A systematic review of 6055 patients with DGS revealed 1) 90.5-100% of patients with advanced imaging of the occiput had at least one occipital-cervical anomaly; 2) Scoliosis was described in 2264 patients; 3) Club foot deformities were the most common described anomaly of the limbs and 4) Lower bone density was also a finding in 22q11.2 deletion [36]. The disorder may be noted from a full blood count by a thrombocytopenia and macrothrombocytopenia (mean platelet volume > 10 fL) [37].
Acute bleeds in congenital haemophilia occur in the hinge joints with the elbow, ankle and knee joints most frequently involved. Blood is highly irritant to the synovium and without appropriate therapy and rehabilitation of the acute hemarthrosis a chronic synovitis is likely to develop [38]. Muscle weakness during a phase of joint immobilization following chronic synovitis is not an uncommon musculoskeletal complication [38].
Skeletal deformity is not only driven by the hemophilic arthropathy alone, but bleeding into musculature with ensuing local ischemia and necrosis may result in contracture formation. When bleeding is into larger muscle group (gluteal region), entrapment neuropathies occur.
Musculoskeletal manifestations of haemophilia are typical in affected males, however it may also occur in a proportion of female carriers. Joint bleeds in female carriers tend to be unrecognized with chronic joint complications being frequent 38.
Patients with haemophilia have a lower bone mineral density compared to the general population which is partly explained by the severity of their chronic illness as well as prolonged periods of joint immobilization due to their haemophilic arthropathy 38. Figure 4 displays typical radiographic findings of a 28-year-old male with radiographic findings typical of haemophilic arthropathy with mild widening of the intercondylar notches (red arrow below) bilaterally (left greater than right). The Figure 4 also displays secondary degenerative changes with joint space narrowing, subchondral sclerosis and left side predominant subchondral cysts.
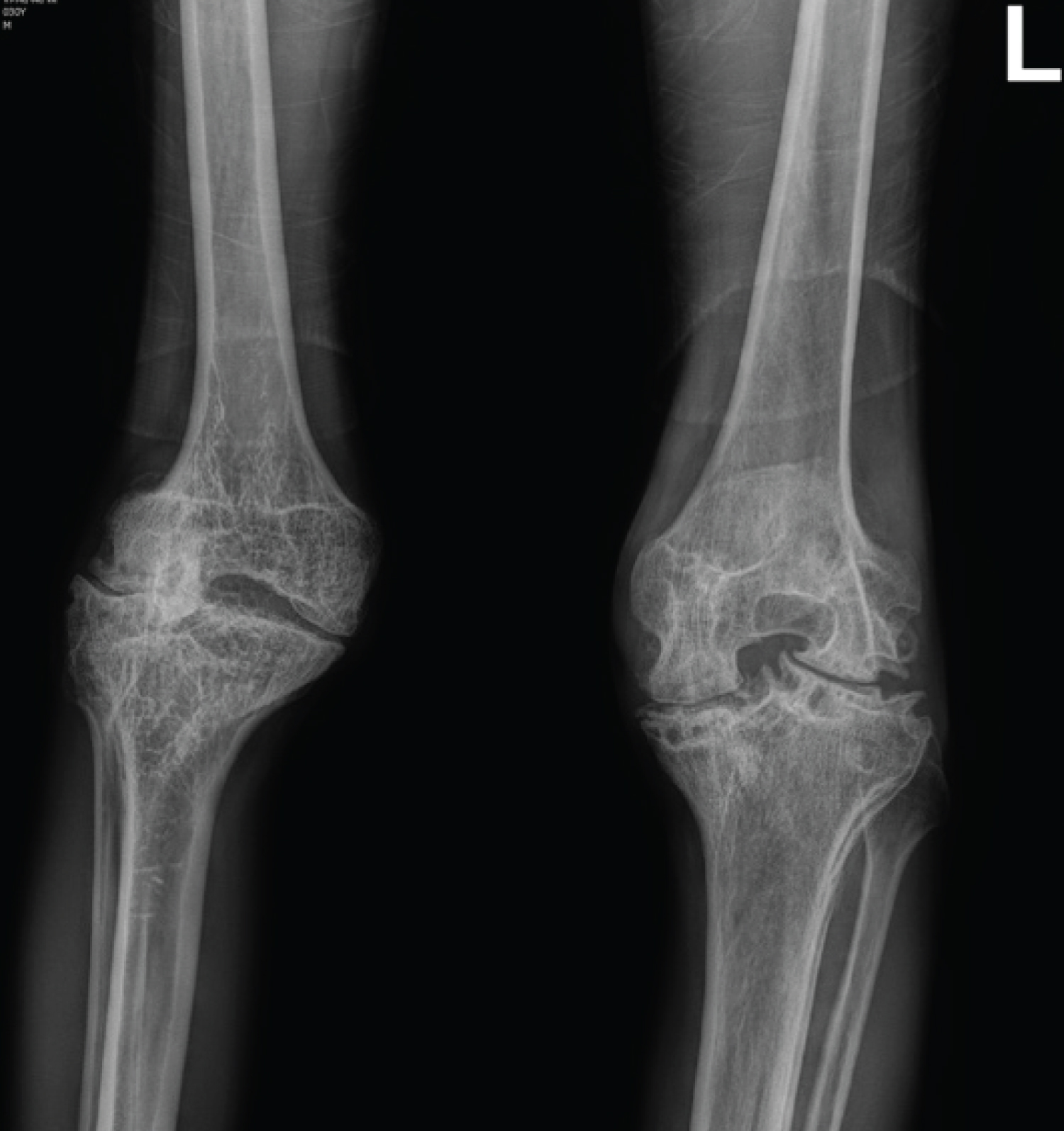 Figure 4: Bilateral knee X-rays of a 28-year-old male with severe haemophilia A and a haemophilic arthropathy (Stage IV disease according to Arnold-Hilgartner classification).
View Figure 4
Figure 4: Bilateral knee X-rays of a 28-year-old male with severe haemophilia A and a haemophilic arthropathy (Stage IV disease according to Arnold-Hilgartner classification).
View Figure 4
A rare coagulation disorder with a reported incidence of between 1.20 million to 1.48 million a year is typified by the presence of neutralizing antibodies (inhibitors) against factor VIII. The pattern of bleeding is variable, and patients may present with mild bleeding in the form of subcutaneous hematomas or life-threatening bleeds [39]. Underlying conditions implicated in the disease pathogenesis include pregnancy and the puerperium, autoimmune conditions, malignancies and infections (hepatitis virus). Among the musculoskeletal presentations in series of 4 patient with AHA at a teaching hospital in Cape Town (South Africa), haematomas in various locations (lower limb, sub-lingual, neck) as well as bilateral knee haemarthroses [40] (Figure 5).
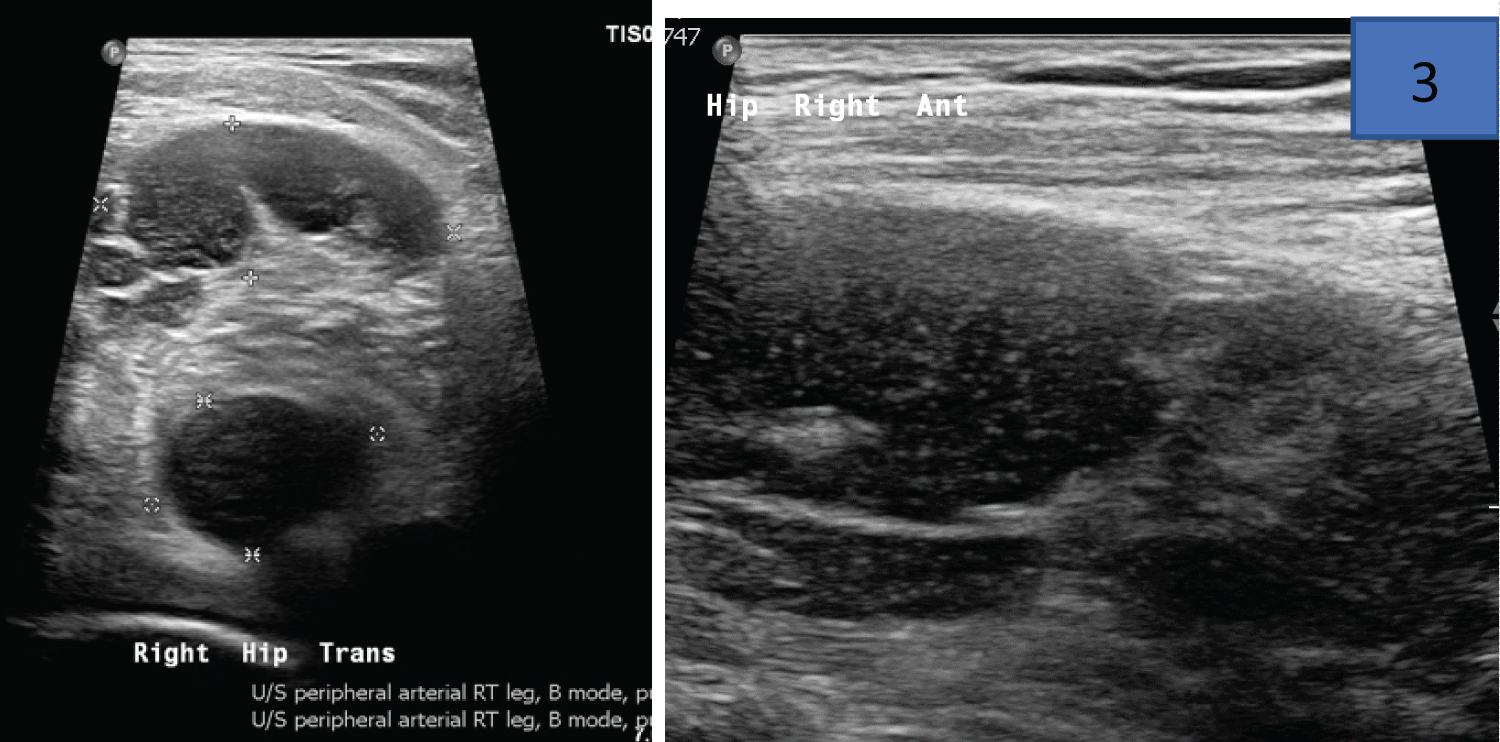 Figure 5: Ultrasound images from a 50-year-old HIV positive male with acquired haemophilia noting a complex right hip collection on transverse and longitudinal imaging of the right hip joint. This patient’s partial thromboplastin time (PTT) was 80.6 seconds at diagnosis with FVIII levels of 0.3 IU/dL.
View Figure 5
Figure 5: Ultrasound images from a 50-year-old HIV positive male with acquired haemophilia noting a complex right hip collection on transverse and longitudinal imaging of the right hip joint. This patient’s partial thromboplastin time (PTT) was 80.6 seconds at diagnosis with FVIII levels of 0.3 IU/dL.
View Figure 5
Glucocorticoids
TKIs: Tyrosine Kinase Inhibitors
Iron Chelators
Bisphosphonates and RANK-L inhibitors
Unfractionated Heparin
Radiation Therapy
Teratogens
Anti-thymocyte globulin and serum Sickness
Glucocorticoids used in supra-physiological doses and for prolonged periods of time eventually result in avascular or ischemic necrosis of cartilage and bone (AVN) 3. Magnetic resonance imaging (MRI) is sensitive in the early detection of osteonecrosis (bone marrow oedema), whilst plain radiographs reveal changes late in the pathologic process when patchy areas of osteopenia and osteosclerosis may be seen 3. Figure 6 findings from a patient with idiopathic immune thrombocytopaenia (ITP) treated with prolonged use of glucocorticosteroids displays typical features of right hip AVN with associated femoral head contour disruption and flattening and joint space narrowing. This patient also had a total left hip replacement for AVN of the left hip.
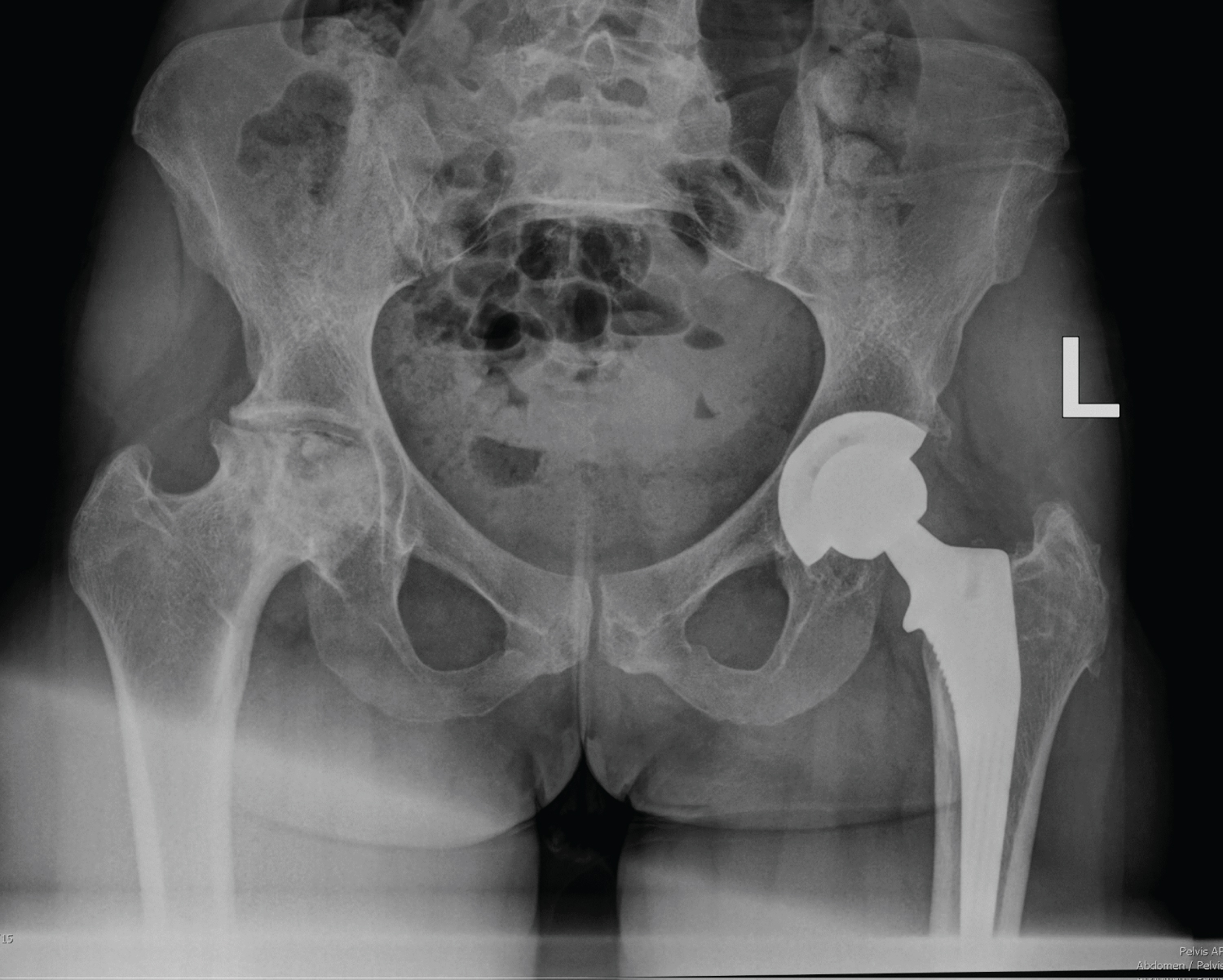 Figure 6: X-Ray findings from an adult patient with right hip avascular necrosis and evidence of a left total hip replacement.
View Figure 6
Figure 6: X-Ray findings from an adult patient with right hip avascular necrosis and evidence of a left total hip replacement.
View Figure 6
Treatment of childhood acute lymphoblastic leukemia (ALL) with chemoimmunotherapy (CIT) relies heavily on glucocorticoid use. High cumulative doses of steroids significantly increase the incidence of osteoporosis and avascular necrosis of bone. Early identification of those at risk for osteoporosis is important so that means to prevent fractures can be considered [41].
Tyrosine kinase inhibitors (TKIs), although associated with significant improvements in survival in CML, have important adverse health risks in children due to their effects on bone growth potential. The side effects of TKIs on endocrine and metabolic organs are mediated through the tyrosine kinase (TK) receptors expressed in these organs and the downstream effector molecules linked to the tyrosine kinase receptors [42]. Use of the first generation TKI (Imatinib mesylate) is postulated to exert effects on the growth hormone/insulin-like growth factor-1 axis (IGF-1) resulting in abnormalities in childhood skeletal growth through its effects on osteoclastogenesis, reduced bone turnover and altered calcium and phosphate metabolism [43]. These deleterious effects on osteoblast and osteoclast activity leads to both osteopenia and osteoporosis in both sexes [44]. Dasatinib, a second-generation TKI, is also noted to have similar deleterious effects on childhood growth potential indicating this as a potential overall class-effect of TKIs on bone metabolism [43]. In the era of chronic myeloid leukemia when treatment free remission (TFR) is sought, trials of tyrosine kinase inhibitor treatment discontinuation may produce a “TKI withdrawal syndrome”. The syndrome is likened to symptoms in polymyalgia rheumatica with joint pain and stiffness reported [45].
The iron chelators are of particular importance in their skeletal manifestations. The parentral agent, DFO: desferrioxamine has particular side effects which include ototoxicity, retinal toxicity, growth defects as well as cartilage and bone abnormalities [46]. Growth retardation is a concern when desferrioxamine (DFO) is used as iron chelation especially when administered in doses greater than 40 mg/Kg/day and when drug therapy is commenced in children under the age of three. Rickets-like bony lesions and genu valgum may be seen in association with radiological changes within the metaphyseal regions of the vertebral bone. Other radiographic changes are vertebral demineralization and flattening of the vertebral bodies [47].
Among the pharmacological agents in the treatment of the SRE: Skeletal Related Events in plasma cell myeloma include the bisphosphonates and monoclonal antibodies targeting RANKL (denosumab). Both of these agents share an uncommon complication of osteonecrosis of the jaw (ONJ) [48]. Bisphosphonate use may also be associated with bone, joint and muscle pain following its administration [48].
The long-term effects of prolonged (> 6 month) use of unfractionated heparin (UFH) has mainly been studied in populations of pregnant women and studies of 237 pregnant patients with prolonged UFH therapy noted a significant reduction in Bone mineral density (BMD) [49].
Radiation therapy is of particular concern in bone owing to its abundant calcium composition. The high calcium content confers increased absorption of radiation, increased local inflammation with accompanying cytokine production predisposing to local damage and fibroatrophy [50].
Use of teratogenic drugs must be undertaken with caution in women of childbearing potential during the course of treatment for their haematological disease. Teratogenic drugs used during critical periods of organogenesis in fetal development are associated with significant musculoskeletal anomalies. Teratogens of notoriety and used within the sphere of haemato-oncology, include thalidomide , warfarin and the vitamin A derivatives.
Thalidomide use during critical periods of organogenesis in foetal development (days 20-36 post-conception) are capable of producing debilitating consequences in the developing foetus with the development of a “thalidomide embryopathy”. Phocomelia is a well-documented skeletal deformity occurring within a spectrum of anomalies in the thalidomide embryopathy and is the result of severe shortening of the limbs due to the reduced length or complete absence of proximal long bones, with only distal elements of the limb remaining [51]. Thalidomide when used in the treatment of adult patients with haematological disease may also be associated with the development of a dose-dependent sensorimotor length-dependent axonal neuropathy [52].
The warfarin-embryopathy is characterised by nasal hypoplasia, shortened limbs and digits (brachydactyly) as well as stippled epiphyses [53]. Post-mortem features of the warfarin-embryopathy are appreciated in Figure 7 [54].
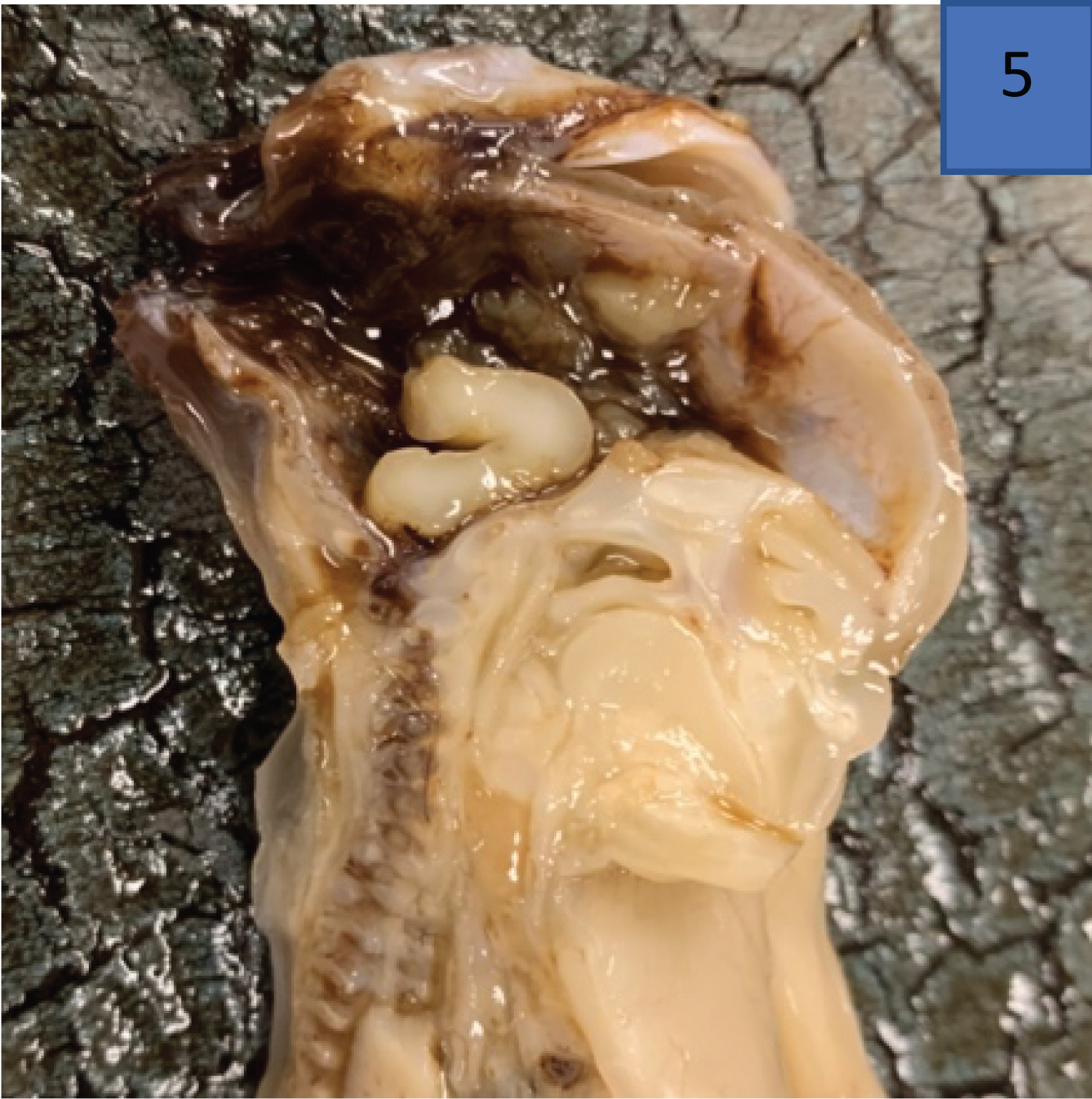 Figure 7: Warfarin embryopathy (anencephaly) demonstrated on post-mortem examination of a male fetus (image used with permission from author Dr Refilwe Monaheng) [54].
View Figure 7
Figure 7: Warfarin embryopathy (anencephaly) demonstrated on post-mortem examination of a male fetus (image used with permission from author Dr Refilwe Monaheng) [54].
View Figure 7
Serum sickness , an immune-complex reaction due to the injection of a foreign protein or proteins, typically occurs one to two weeks post exposure and is documented following exposure to anti-thymocyte globulin infusions as part of Immune Suppressive Therapy (IST) in aplastic anaemia. The illness presents with fever, a urticarial rash with pain and swelling of joints in the knees, ankles and wrists [55].
Plasma cell neoplasms, light chain amyloidosis and POEMS syndrome
Lymphoma: Hodgkin and non-Hodgkin
Hodgkin: Classical Hodgkin and Nodular lymphocyte predominant
Non-Hodgkin: B and T-cell
Myeloproliferative Neoplasms
There exists a close relationship between rheumatological conditions and the development of clonal haematological disorders. Meta-analyses have demonstrated that non-Hodgkin lymphoma NHL is more common in patients with autoimmune diseases like Primary Sjogren Syndrome (PSS) and Systemic Lupus Erythematosus (SLE). Factors in rheumatoid arthritis (RA) that may explain the development of lymphoma are the depth and duration of inflammation-with longer standing and more profound inflammation accounting for the risk of developing aggressive large B-cell lymphomas. The extent to which cytotoxic therapies contribute to lymphomagenesis is noted in rheumatoid arthritis, with risk of NHL being appreciably greater with the use of cytotoxic or biologics compared to conventional anti-rheumatic treatment alone. The association with indolent mucosa associated lymphomas is strongly associated with PSS [56]. The central role of inflammation in lymphomagenesis is recognized in T-cell large granular lymphocytic leukemia (T-LGL) in which in which 25-35% of patients are noted to have co-existing rheumatoid arthritis (RA) or another auto-immune disease [57].
Plasma Cell Myeloma (PCM) is typified by clonal proliferation of bone marrow plasma cells with a heterogeneous degree of either disseminated bone marrow involvement or multi-focal marrow involvement. The WHO diagnostic classification of tumours of haematopoietic and lymphoid tissues criteria for diagnosing PCM includes evidence of ≥ 1 osteolytic lesion visualized by plain radiograph, computerized tomography (CT) or PET/CT) and > 1 focal lesion visualized on Magnetic Resonance Imaging (MRI) [58]. Bone pain is a frequent complication of PCM and may be ascribed to lytic lesions or pathological vertebral fractures. Pain was a presenting symptom in 94.1% of patients diagnosed with PCM at a tertiary hospital in Soweto (Johannesburg, South Africa) over a six-year period [59].
The costo-iliac height (Figure 3), that height between the lower costal margin and iliac crest, is demonstrated below. The costo-iliac height, also referred to as Seftels sign after Professor Harry Seftel (personal communication from emeritus Professor, Moosa Patel), is often altered in patients with multiple myeloma and significant loss of the lumbar vertebral height. The usual height of two-finger breadths is reduced as the two surfaces become apposed or overlap with alterations in lumbar vertebral height, which may occur due to vertebral body fracture/collapse in patients with multiple myeloma [60] (Figure 8).
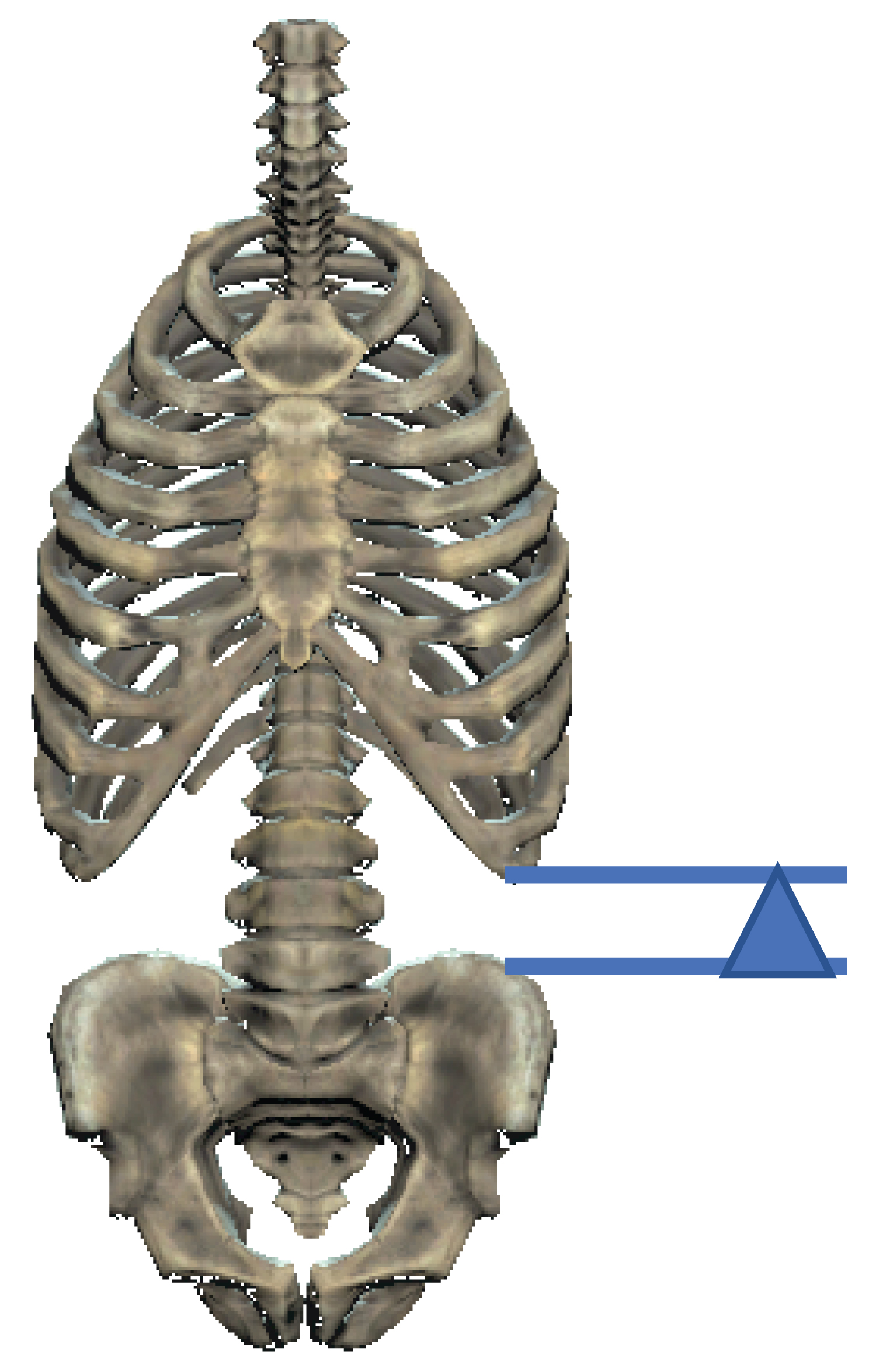 Figure 8: The costo-iliac height (arrow-head) 60.
View Figure 8
Figure 8: The costo-iliac height (arrow-head) 60.
View Figure 8
Patients with solitary plasmacytomas frequently present with localized bone pain or a pathological fracture. Vertebral lesions may be associated with symptomatic cord compression and soft tissue extension may produce a palpable extra-dural mass lesion [58].
Within the disease entity plasma cell leukemia , lytic bone lesions and bone pain occur but are seen less frequently than plasma cell myeloma. In primary plasma cell leukemia, extramedullary plasmacytomas are more likely to occur than in secondary cases of plasma cell leukemia. In secondary plasma cell leukemia, advanced bone disease is more frequently noted [58].
Patients with post-transplant lymphoproliferative neoplasms with a myeloma-like presentation may present with osteolytic skeletal lesions [61].
Monoclonal gammopathy of undetermined significance (MGUS) and smoldering myeloma (SMM) are regarded as pre-malignant conditions with a risk of progression to myeloma at 1% annually in the former and 10% in the first five years in the latter. MGUS and SMM both have an increased risk of osteoporosis and bone fractures even in the absence of progression to overt lymphoplasmacytic malignancy, with peripheral neuropathies also not uncommon in MGUS [62] (Figure 9).
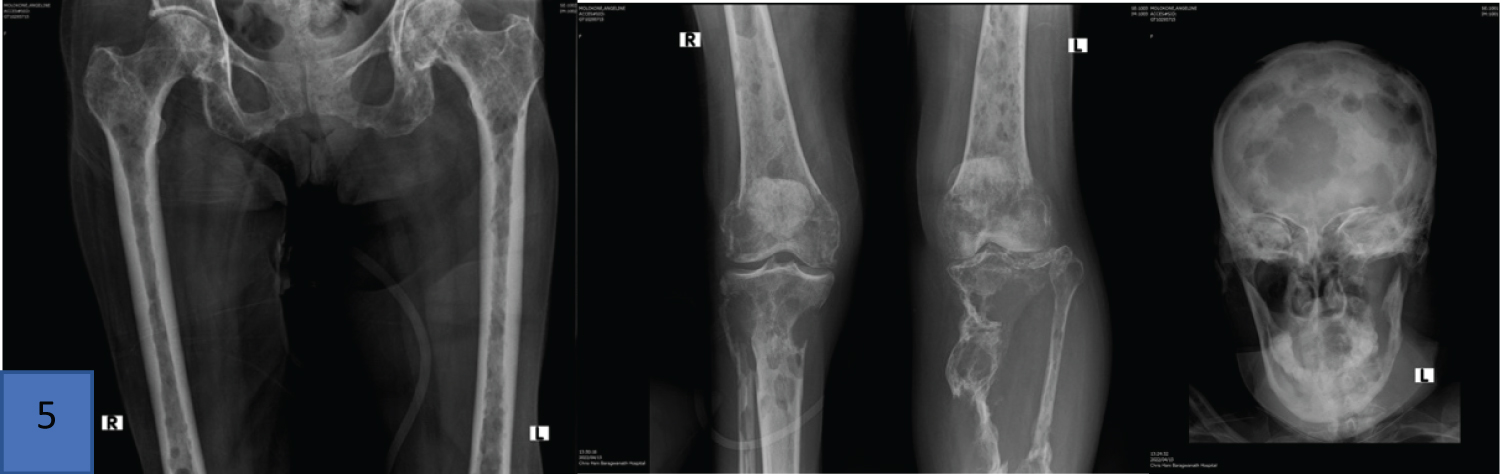 Figure 9: Images from a skeletal survey of a 68-year-old patient diagnosed with IgG Kappa plasma cell myeloma noting multiple lytic lesions with associated left tibial and fibula pathological fractures.
View Figure 9
Figure 9: Images from a skeletal survey of a 68-year-old patient diagnosed with IgG Kappa plasma cell myeloma noting multiple lytic lesions with associated left tibial and fibula pathological fractures.
View Figure 9
The Figure 10 of a 53-year-old male with relapsed/refractory multiple myeloma with multiple lines of prior therapy and a prior autologous stem cell transplant (ASCT). At relapse, following bortezomib based therapy, there was extensive disease noted clinically with multiple extra-medullary plasmacytomas over the forehead and within the axillae. Neurological examination revealed patchy sensory loss with motor weakness of L1/L2 dermatome. MRI of the spine showed an intrathecal soft tissue tumour at T3 and T5 level encasing the spinal cord.
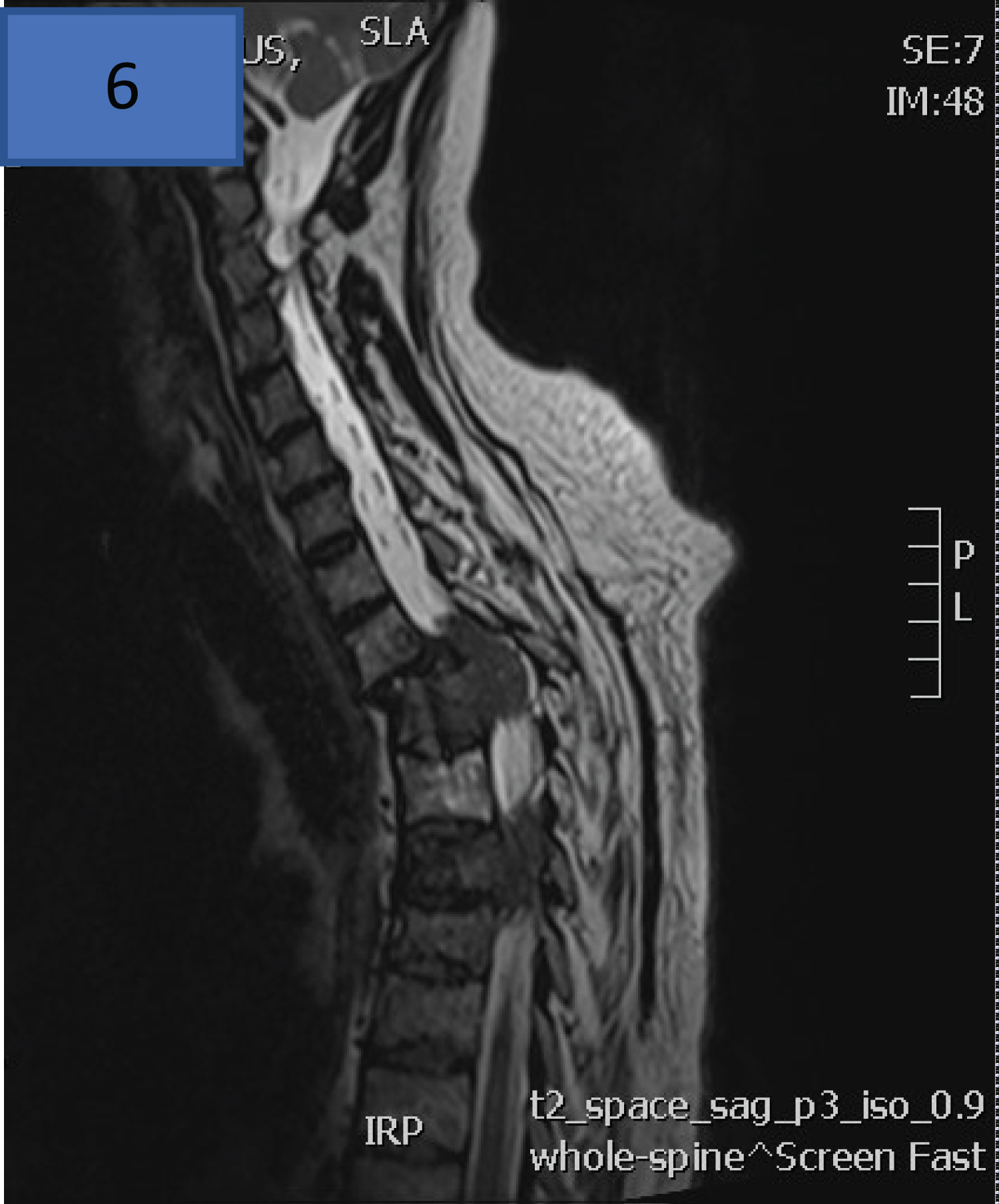 Figure 10: MRI Spine of a 53-year-old male with relapsed/refractory plasma cell myeloma with an intra-thecal soft tissue tumour at T3 and T5 level encasing the spinal cord.
View Figure 10
Figure 10: MRI Spine of a 53-year-old male with relapsed/refractory plasma cell myeloma with an intra-thecal soft tissue tumour at T3 and T5 level encasing the spinal cord.
View Figure 10
Systemic light chain amyloidosis (AL), due to a monoclonal paraproteinemia encountered with plasma cell myeloma and Waldenstroms macroglobulinemia, has varied musculoskeletal manifestations including the development of myopathies, pseudohypertrophy of joints (glenohumeral joints), arthropathies and bony lesions [63].
Soft tissue swelling in AL may be prominent in up to 75 percent of cases due to a nodular hypertrophied synovium directly infiltrated by amyloid. Swelling may be particularly prominent around the glenohumeral joint, resulting in a characteristic "shoulder pad" sign [63]. Subcutaneous nodules in amyloid are differentiated from rheumatoid nodules, by Congo red staining with amyloid demonstrating birefringence when viewed under polarised light [63].
POEMS syndrome is encumbent of the features of the acronym polyradiculoneuropathy (P), organomegaly (O), endocrinopathy (E), a monoclonal plasma cell dyscrasia (M) with skin changes (S). A further mnemonic, PEST summarizes other encountered features including: Papilloedema (P), extra-vascular volume overload (E), sclerotic bone lesions (S) and thrombocytosis (t) [64]. The osteosclerotic lesions occur in 95% of cases and vary from single sclerotic lesions in half of patients to more than three lesions in up to a third of patients [58].
Lymphomatous involvement of bone as a primary occurrence is relatively infrequent and generally represents secondary bony involvement in the form of extranodal disease [65]. The occurrence of extranodal disease is not infrequent in non-Hodgkin lymphoma with one-third of non-Hodgkin lymphomas (NHL) presenting with extra-nodal or extra-medullary disease. Extra-nodal sites that are frequently involved include the gastrointestinal tract, head and neck, lung, skin, bone and brain [66]. The co-occurrence of HIV alters the disease presentation in NHL, with patients presenting at a significantly younger age with more aggressive histology, greater extranodal disease and a poorer prognosis [67,68].
Hodgkin lymphoma: The classical description of Hodgkin lymphoma (HL) at presentation is that of a disease involving lymph node stations above the diaphragm with a “predictable” contiguous spread via the lymphatics. Extra-nodal (EN) disease in HL is less common than in non-Hodgkin lymphoma (NHL) with bony extra-lymphatic spread regarded as uncommon in HL 69. Osseous involvement in HL has been described radiologically as osteosclerotic, osteolytic, or with a mixed osteosclerotic/osteolytic picture 69. Hodgkin lymphoma in the setting of HIV infection has a more aggressive clinical course with bone marrow involvement representing a common extra-nodal site 69.
The post-contrast magnetic resonance image (MRI) Figure 11 represents the often atypical and advanced presentation of a 22-year-old male, HIV negative, biopsy proven stage IV Hodgkin lymphoma. Imaging (MRI) displayed a large right anterior paraspinal mass with avid enhancement (T5-T9) complicated by multilevel neuroforamina extension and marked cord compression.
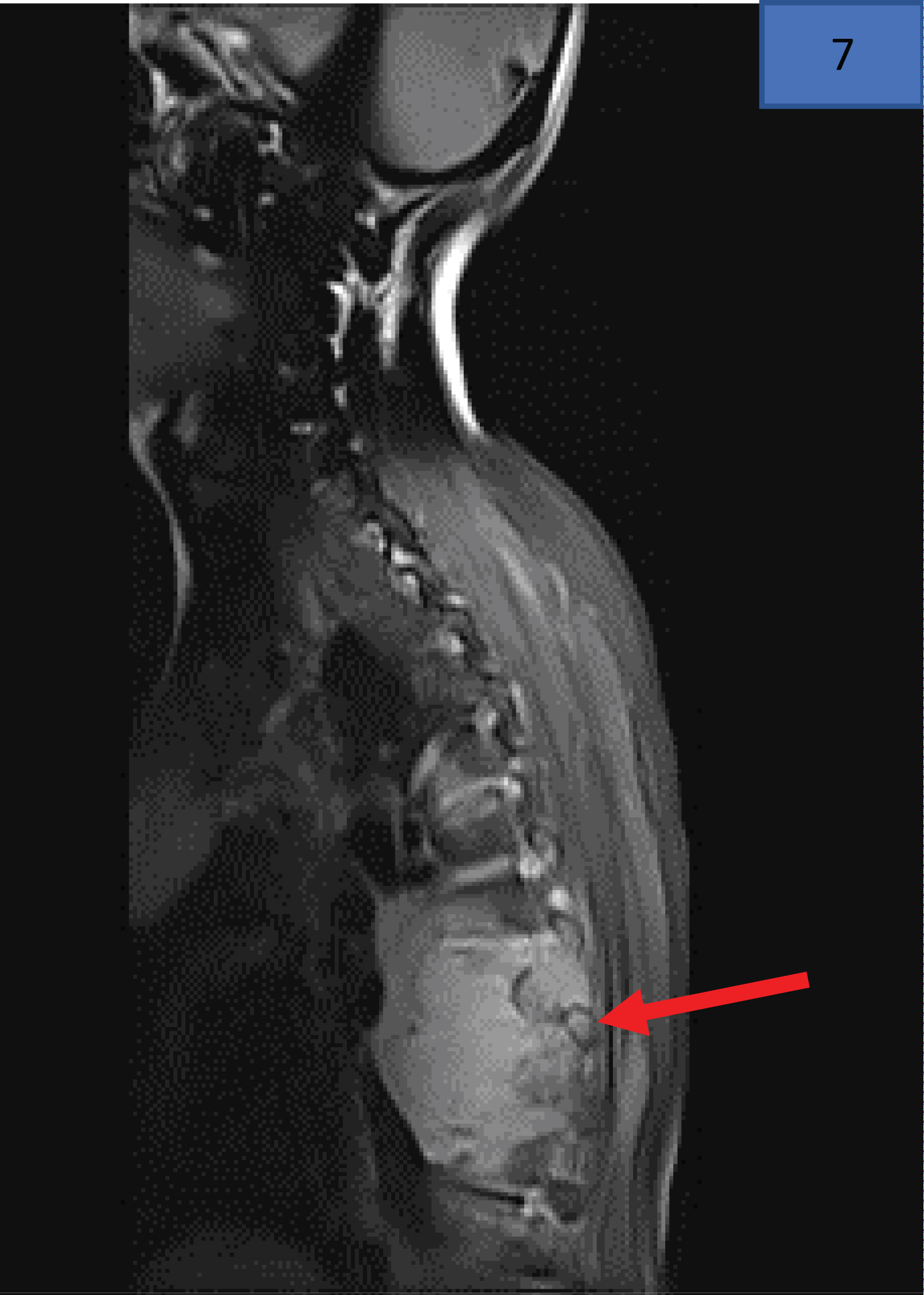 Figure 11: MRI findings of a 22-year-old male, diagnosed with stage IV Hodgkin Lymphoma, who presented with presented with 8-month history of lower back pain and an acute history of neurological fallout in the lower limbs (sensory level, bladder/bowel involvement). The red arrow indicates the paraspinal mass infiltrating the vertebra.
View Figure 11
Figure 11: MRI findings of a 22-year-old male, diagnosed with stage IV Hodgkin Lymphoma, who presented with presented with 8-month history of lower back pain and an acute history of neurological fallout in the lower limbs (sensory level, bladder/bowel involvement). The red arrow indicates the paraspinal mass infiltrating the vertebra.
View Figure 11
Rare cases of patients with advanced stage nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) may also have destructive lesions involving the bone [70].
Non-Hodgkin lymphomas: B and T-cell: Primary non-Hodgkin lymphoma of bone is rare and is defined as malignant lymphoid neoplasm producing at least one bony mass lesion bone without the involvement of extra-regional lymph nodes and extra-nodal sites and documented for a period of six months. Any part of the skeleton may be involved with lesions of the femur and humerus more frequently reported. Disease epidemiology reveals a male preponderance with mean age of 48 years. The immunophenotype of primary NHL of bone is predominantly B-cell with admixed reactive T-cell infiltrates commonly co-occurring [71]. Patients with primary diffuse large B-cell lymphoma of bone often present with pain and swelling or pathologic fracture of the affected area of the skeleton [72].
The Figure 12 of a 57-year-old female, HIV positive, with a pathological right femur fracture with adjacent large soft tissue mass. Intra-operative biopsy findings revealed a diffuse large B-cell lymphoma. This patient received multi-agent chemotherapy as well as placement of an intra-medullary nail.
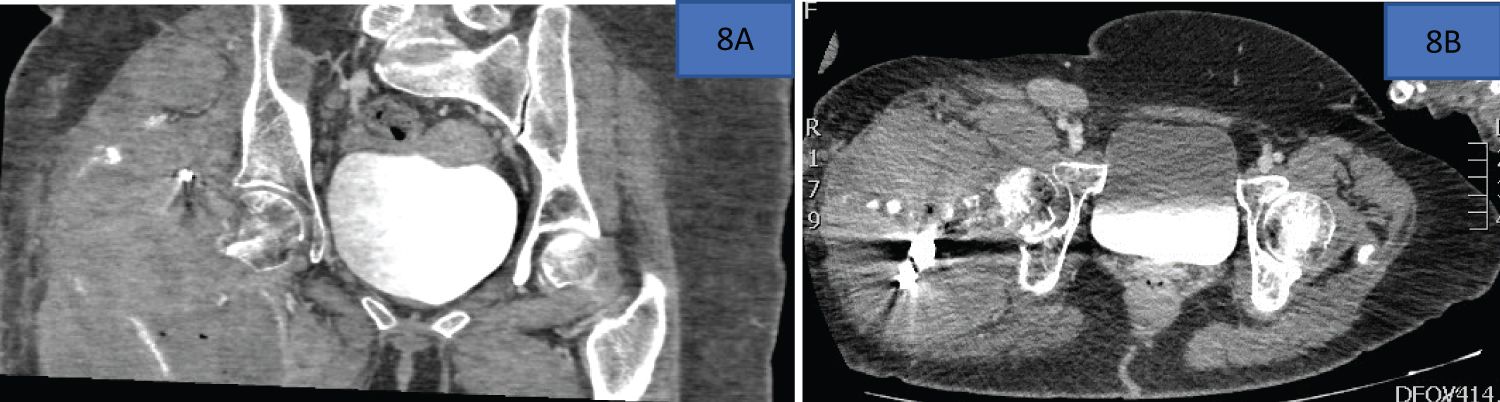 Figure 12: Computerized tomography images of a patient with a pathological right femur fracture and right leg adjacent soft tissue mass.
View Figure 12
Figure 12: Computerized tomography images of a patient with a pathological right femur fracture and right leg adjacent soft tissue mass.
View Figure 12
Plasmablastic Lymphoma (PBL), an aggressive B-cell non-Hodgkin lymphoma, occurring more typically in the setting of immune compromise (iatrogenic, acquired, autoimmune) has the propensity to involve extra nodal sites with often devastating local disease in the head and neck [73]. Data from Rapiti, et al. (Kwazulu natal, UKZN) noted that all of the patients (n = 26) in their cohort presented with extra-nodal disease with lytic bone lesions noted in 5 patients [74]. The presence of lytic bone lesions in PBL raises a diagnostic concern when differentiating it from plasma cell myeloma [73]. Figure 13 is a volume-rendered image from the CT-scan of a 27-year-old male with HIV, diagnosed with plasmablastic lymphoma, showing an oral exophytic lesion of histologically confirmed plasmablastic lymphoma.
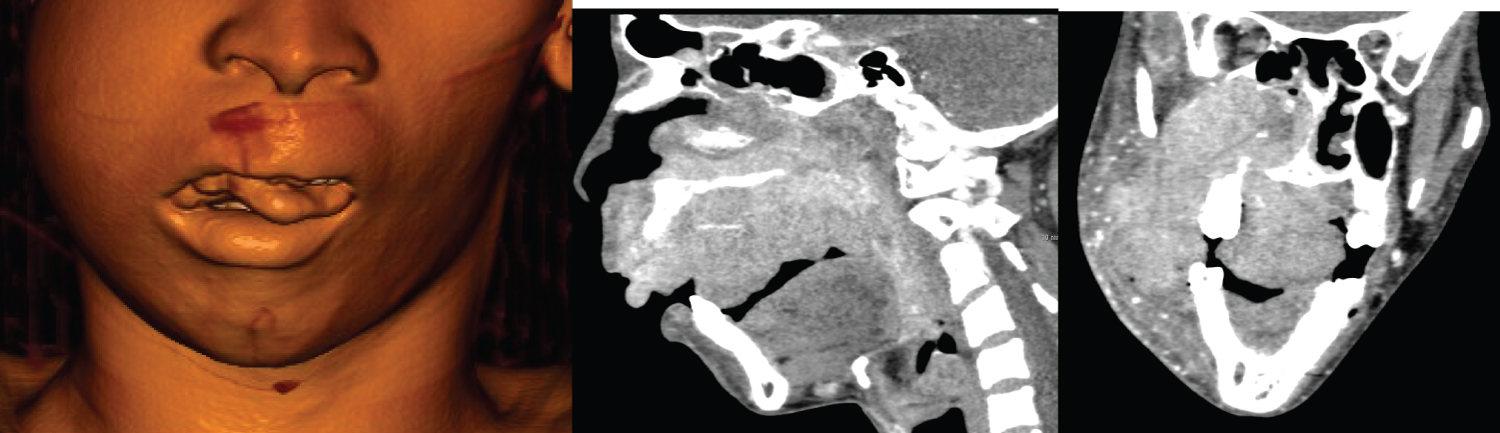 Figure 13: Coronal and Sagittal CT images of a 27-year-old male diagnosed with plasmablastic lymphoma a,b) With imaging displaying a heterogeneously enhancing expansile mass involving the paranasal sinuses and oral cavity with bony erosion of right maxillary sinus and c) Right orbital floor (red arrow head).
View Figure 13
Figure 13: Coronal and Sagittal CT images of a 27-year-old male diagnosed with plasmablastic lymphoma a,b) With imaging displaying a heterogeneously enhancing expansile mass involving the paranasal sinuses and oral cavity with bony erosion of right maxillary sinus and c) Right orbital floor (red arrow head).
View Figure 13
T-cell non-Hodgkin lymphoma
Adult T-Cell Leukemia-Lymphoma (ATLL) is a mature T-cell non-Hodgkin lymphoma that epidemiologically tracks areas with high HTLV endemicity [75]. The disease entity is subdivided into four subtypes with the relative frequency of hypercalcemia and lytic bone lesions differing between the subtypes. Patients with the acute variant will invariably develop hypercalcemia at some point in the natural history of the disease with close to half of patients developing lytic bone lesions [75]. Patients with hypercalcemia in ATLL often have FDG-PET scan positive results at sites of bony lesions. Lytic bone lesions are linked to the production of parathyroid hormone-related protein by tumour cells [57]. Data from a epidemiological study in South Africa noted that over the ten year study period noted a disease incidence of 0.06 per 100,000 population with the bulk of cases falling in the acute subtype [76].
The presence of bone lesions in anaplastic large cell lymphoma is described radiologically as osteolytic bone lesions with involvement of multiple sites [77]. Figure 14 demonstrates the locally aggressive nature of anaplastic large cell lymphoma in a patient with a forehead mass with Figure 15 noting significant facial asymmetry produced by an aggressive NK/T-cell lymphoma in an HIV-negative patient [78].
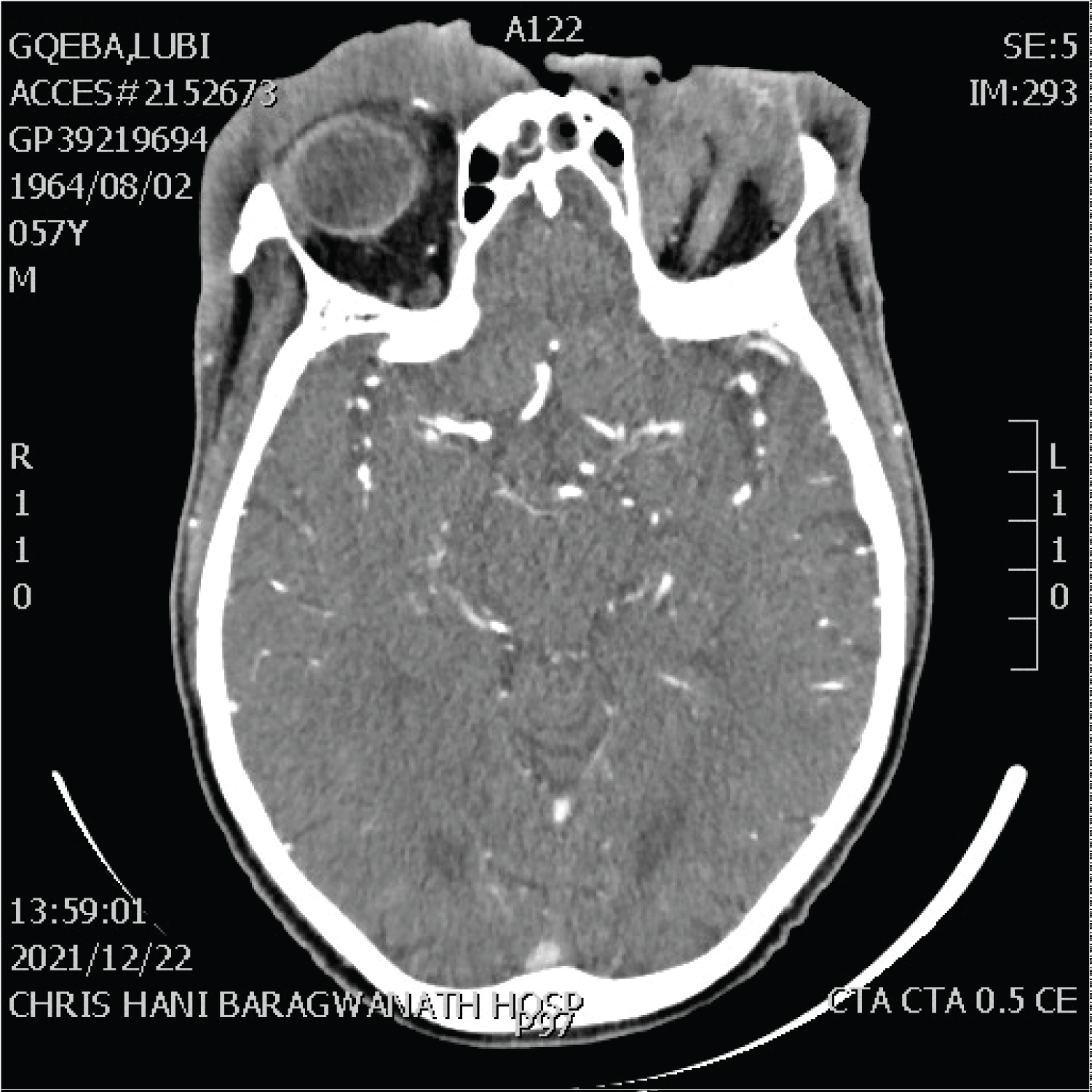 Figure 14: 56-year-old male, HIV negative, diagnosed with anaplastic large cell lymphoma presenting as an ulcerative and locally destructive lesion of the left-eye (with both pre and post septal involvement) and forehead, crossing the midline to involve the right pre-septal soft tissues.
View Figure 14
Figure 14: 56-year-old male, HIV negative, diagnosed with anaplastic large cell lymphoma presenting as an ulcerative and locally destructive lesion of the left-eye (with both pre and post septal involvement) and forehead, crossing the midline to involve the right pre-septal soft tissues.
View Figure 14
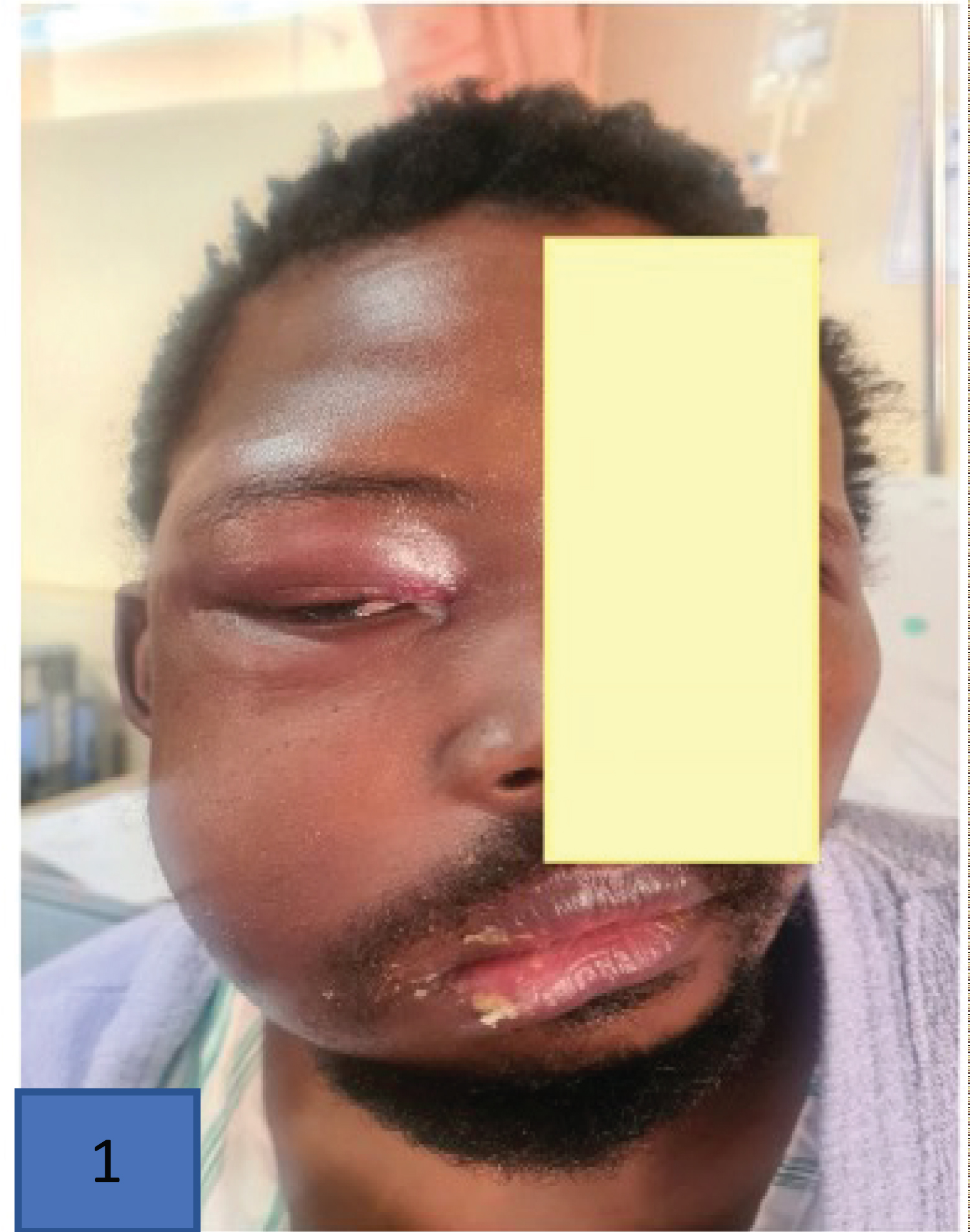 Figure 15: Facial asymmetry in a 31-year-old male, HIV negative, with histological diagnosis of extranodal natural killer (NK)/T-cell lymphoma nasal type [78] (image used with permission from cited publication by author Professor Moosa Patel).
View Figure 15
Figure 15: Facial asymmetry in a 31-year-old male, HIV negative, with histological diagnosis of extranodal natural killer (NK)/T-cell lymphoma nasal type [78] (image used with permission from cited publication by author Professor Moosa Patel).
View Figure 15
Myeloid sarcomas , also known as granulocytic sarcomas or chloromas, are tumor masses consisting of myeloid blasts which occur at sites other than the bone marrow. They may present at initial diagnosis of an acute leukemia or may represent the only finding of relapsed disease [79]. Cytogenetic alterations in AML associated with granulocytic sarcomas include t (9;11) (p21.3;q23.3), inv(16)(p13.1q22) or t(16;16)(p13.1;q22) [79]. Frequently involved sites include skin, lymph nodes, gastrointestinal tract, bone, soft tissue and the testes.
In primary myelofibrosis (PMF) and polycythemia vera (PCV), an increase in cell turnover may precipitate hyperuricemia as well as gouty arthritis [80]. Patchy or diffuse osteosclerosis in PMF is a common finding, as are “sandwich vertebrae,” so called because of marked radiodensity of superior and inferior margins of the vertebral body. MRI can identify the uncommon periosteal reactions that usually occur in the distal femur, proximal tibia, or ankle. The reactions represent expansion of marrow cellularity into normally inactive regions of long bones or extramedullary space-occupying lesions of fibrohematopoietic tissue [80].
EMH: Extra-Medullary Haemopoiesis, is a physiological and compensatory attempt to produce blood cells in the face of cytopenias in PMF. EMH is either hepatosplenic or non-hepatosplenic, in the latter involving the central nervous system as epidural soft tissue masses [81]. In instances where there is spinal cord compression with neurological fallout involved field radiation therapy is the preferred emergency therapy [82]. Osteosclerosis occurs in 30-70% of cases with periosteal reactions occurring at the metaphyses of the distal femur and proximal tibia [18]. F-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) in PMF characteristically demonstrates intense and diffuse tracer uptake both in osteosclerotic bones and extraosseous sites of EMH [81].
One of the presenting features of systemic mastocytosis is the involvement of the musculoskeletal system with bone pain, osteopenia, osteoporosis, fractures as well as myalgia and arthralgias. Mast cell sarcomas, albeit rare, have the potential to form localized masses with a destructive growth pattern [83].
Histiocytic disorders
Haematological disease and scleroderma-like skin changes
Parvovirus B19: pure red cell aplasia and arthropathy
Haematology and the nervous system
Haematology and somatic/dermatological disease
Over 100 histiocytic disorders have been described in literature with classification systems dividing them into 5 classes: 1) L-group including Erdheim-Chester disease and Langerhans cell histiocytosis as examples (LCH), 2) C-group (cutaneous non-LCH and cutaneous non-LCH with major systemic response,3) R-group (Familial and sporadic Rosai-Dorfmann disease), 4) M-group (primary and secondary malignant histiocytoses) and 5) H-group (primary and secondary histiocytoses) [84].
Langerhans cell histiocytosis presents in over 80% of cases with skeletal lesions of the skull, spine, limbs, and pelvis have been described. Imaging of bones shows lytic lesions without marginal sclerosis with or without periosteal reactions [84].
Involvement of the skeleton occurs in up to 96% of patients with Erdheim-Chester disease (ECD) with bone pain occurring in only half of all cases. Frequently affected bones include the femur, tibia and fibula with less commonly involved sites being the ulna, radius and humerus. The halmark of skeletal involvement in ECD is osteosclerosis with mixed sclerotic and lytic lesions described on occasion [85].
Sclerotic skin lesions resembling systemic scleroderma (SSc) that occur in patients with malignant tumours and other diseases are well described by the terms scleroderma-like, pseudoscleroderma or pseudosclerosis [86].
Scleromyxedema or papular mucionosis is described with systemic light chain amyloidosis (AL) as well as in plasma cell myeloma. Scleromyxedema is characterized by waxy, yellow-red papules on the head, neck, arms and upper trunk and occurs over thickened and indurated skin. Systemic light chain amyloid (AL) and its multi-system involvement incites amyloid depisition not only in critical organs like the heart and kidneys but also in the skin producing thickening and accompanying joint stiffness [63]. AL amyloid affects peripheral nerves producing a symmetrical lower extremity sensorimotor polyneuropathy or symptoms of carpal tunnel syndrome in the hands [87].
Graft versus host disease (GVHD), is one of the feared complications of bone marrow transplantation beyond the acute period of neutropenic sepsis and the concern of delayed engraftment. Skin thickening and scleroderma-like skin changes with joint involvement are a concern in chronic GVHD . Although typically occurrence of these skin changes parallels ASCT, it may even occur following blood product transfusion in immunocompromised hosts [88].
The hallmark of the cutaneous involvement associated with the toxicity of the chemotherapeutic agent bleomycin , is the “flagellate erythematous hyperpigmentation” with sclerodermoid lesions. While these lesions are rarely reported in large-volume centres for lymphoma treatment, the pulmonary toxicities tend to be more frequently recognised with more serious complications [89].
Infection with Parvovirus B19 produces mild disease in children, with the disease in middle-aged women producing significant arthropathy. The resultant arthropathy is a true inflammatory arthritis occurring symmetrically in joints of the hands, ankles, knees and wrists [90]. Both acute and chronic parvovirus infection produce alterations in bone marrow erythropoiesis, with a transient aplastic crisis occurring acutely, with chronic parvovirus infection producing a pure red cell aplasia (PRCA) [90].
Peripheral neuropathies in the setting of haematological disease produce alterations in sensory, motor, and autonomic components of nerves producing pain, altered sensation, functional disability, and increased risk of falls. Drugs implicated in the development of neuropathies are vincristine, thalidomide (described above) and the proteosome inhibitor bortezomib. Disease processes with paraprotein production (plasma cell myeloma, amyloidosis, Waldenstroms macroglobulinemia) are also risk factors for neuropathy development [91].
Deficiency of vitamin B12 not only has profound influence on haematopoiesis (megaloblastic anaemia) but is also an essential component of neural tube development in-utero with neural tube defects (NTD) reported in infants born to mothers with low vitamin B12 levels [92]. Subacute combined degeneration of the cord is a feared consequence of vitamin B12 deficiency with peripheral neuropathies, neuropsychiatric aberrations and optic nerve atrophy described [93].
Hodgkin lymphoma may present with intractable pruritus, with icythosis and erythema nodosum also described in the disease [94]. Direct or primary skin involvement with Hodgkin lymphoma is rarely reported and highlighted in Figure 16 [95].
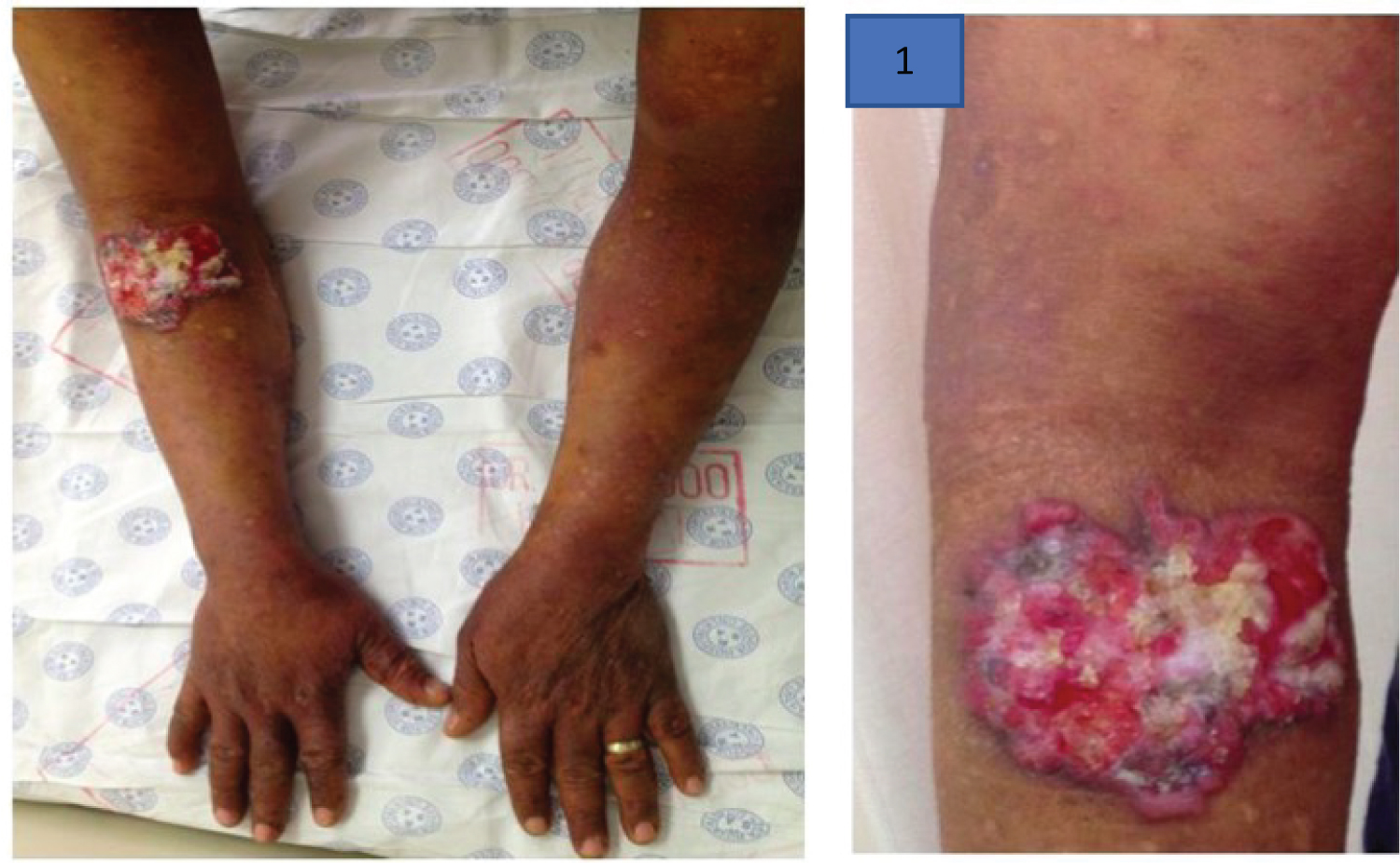 Figure 16: Cutaneous Hodgkin lymphoma [95] (image used with permission from cited article by the co-author Professor Moosa Patel).
View Figure 16
Figure 16: Cutaneous Hodgkin lymphoma [95] (image used with permission from cited article by the co-author Professor Moosa Patel).
View Figure 16
Leukemia cutis occurs when malignant leucocytes infiltrate the skin to producing plaques or nodules. Leukemic deposits occur frequently in acute myeloid leukemia (AML) and similarly have been reported with multiple myeloma [94]. Rare dermatological complications in PCM occur when xanthoma occur diffusely in normo-lipemic patients due to paraprotein-lipoprotein complex deposition in the skin [94]. Figure 16 represents eruptive xanthoma in a case of hyperlipidemic myeloma, a rare form of acquired dysbetalipoproteinemia, in an HIV positive patient with myeloma [96] (Figure 17).
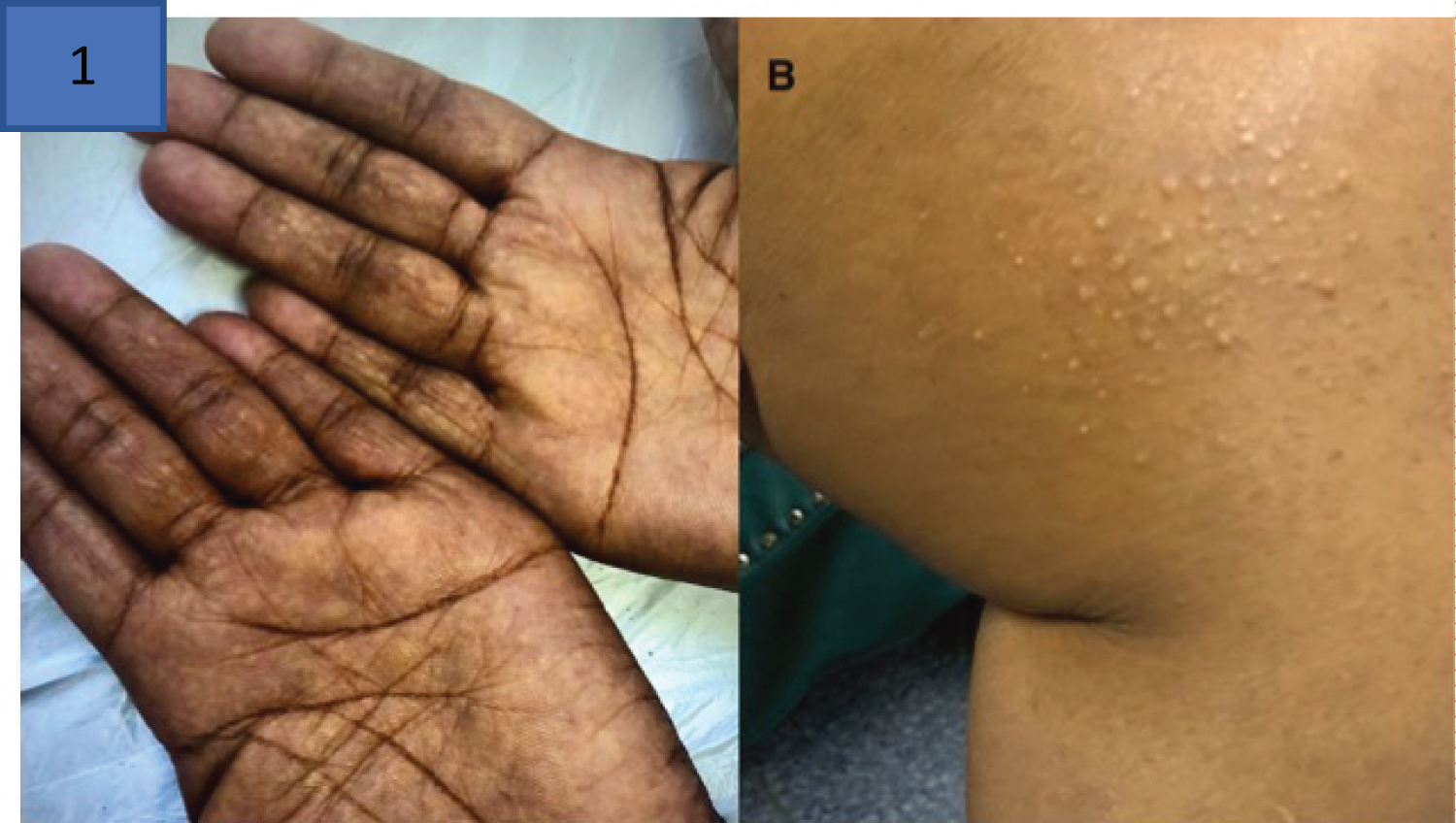 Figure 17: Hyperlipidemic myeloma [95] with a) Palmar xanthoma and b) Eruptive xanthoma on medial aspect of the thigh above the left knee (image used with permission from cited co-author Professor Moosa Patel).
View Figure 17
Figure 17: Hyperlipidemic myeloma [95] with a) Palmar xanthoma and b) Eruptive xanthoma on medial aspect of the thigh above the left knee (image used with permission from cited co-author Professor Moosa Patel).
View Figure 17