Hereby, we present the case of a 54-year-old woman with a myeloid neoplasm associated with ETV6-PDGFRB rearrangement. Clinical examination revealed splenomegaly and hypereosinophilia was observed in peripheral blood (44.8 × 109 WBC/L; eosinophils: 2.24 × 103/mm3). Bone marrow aspirate and biopsy were hypercellular, mainly composed of granular cells with an increased medullary eosinophilia and displayed dysplastic features. Finally, bone marrow cytogenetics showed a t(5;12)(q33;p13) karyotype in 94% metaphases with fluorescence in-situ hybridization (FISH) analysis. Since myeloid neoplasms associated with PDGFRB rearrangements are generally well sensitive to tyrosine kinase inhibitor, an imatinib therapy was initiated (400 mg/daily). Patient responded well to treatment and eleven months after initiation of chemotherapy, patient was still in hematological and cytogenetic remission.
In addition, several uncertainties related to this neoplasm were reviewed in the literature such as male bias, eosinophilic commitment, optimal imatinib dosage regimen, delayed-diagnostic, natural course of the disease (and associated treatment resistance) or imatinib withdrawal in case of deep molecular response.
PDGFRB, ETV6, ETV6-PDGFRB rearrangement, Eosinophilic leukemia, Myeloid neoplasm
The upper limit of normal range of eosinophils in the peripheral blood is an absolute eosinophil count of 350-500/mm3. Hypereosinophilia can further be classified as mild, moderate or severe according to absolute eosinophils counts (up to 1500/mm3, 1500-5000/mm3 and superior to 5000/mm3 respectively). The first step in interpreting hypereosinophilia is to exclude a reactive eosinophilia which can be caused by a variety of secondary conditions. After a cautious exclusion of those, primary eosinophilia can ultimately be considered [1,2].
The World Health Organization (WHO) classification of tumours of hematopoietic and lymphoid tissues, revised in 2008 (and updated in 2016), subclassifies primary eosinophilic disorders based on cytogenetic/molecular and histopathological findings. According to this classification scheme, primary eosinophilic disorders could be classified as chronic eosinophilic leukemia, not otherwise specified (CEL, NOS), Hypereosinophilic Syndrome (HES), idiopathic hypereosinophilia, Lymphocytic Variant Hypereosinophilia (L-HES) or eosinophilia associated with abnormalities of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2 [1-3].
Herein, we report on the case of a 54-year-old woman referred to our institution after the discovery of a hyperleukocytosis during a myocardial ischemia follow-up. Conventional cytogenetics showed a t(5;12)(q33;p13) karyotype in 94% metaphases with Fluorescence In-Situ Hybridization (FISH) analysis confirming the presence of a ETV6-PDGFRB fusion gene. A therapy with imatinib was proposed.
We thus report the case of a patient diagnosed with a myeloproliferative neoplasm associated with eosinophilia and rearrangement of PDGFRB with a good response to an imatinib therapy.
A 54-year-old woman was referred to our department of Hematology in August 2015 with a hyperleukocytosis discovered during a myocardial ischemia follow-up. As a matter of fact, the patient suffered from an inferior ST-segment elevation myocardial infarction the year before presentation. Clinical examination revealed splenomegaly palpable 2 cm below the left costal margin. Hematological data in peripheral blood were 44.8 × 109 WBC/L (neutrophils: 26.4 × 103/mm3, lymphocytes: 6.3 × 103/mm3, monocytes: 2.24 103/mm3, eosinophils: 2.24 × 103/mm3, basophils: 0.5 × 103/mm3, metamyelocytes: 2.7 × 103/mm3, myelocytes: 3.1 × 103/mm3 and promyelocytes: 1.35 × 103/mm3), 236 × 109 platelets/L and an hemoglobin level of 104 g/L (Advia2120®, Siemens, Germany). Bone marrow aspirate was hypercellular and mainly composed of granular cells including 21.8% of eosinophils, 37% of neutrophils, 1.8% of basophils and 24.7% of immature granulocytic cells (blasts = 1.3%, promyelocytes = 3.9%, myelocytes = 7.4%, metamyelocytes = 12.1%). Dysmegakaryopoiesis (Hypo-lobulated megakaryocytes, micromegakaryocytes, karyorrhexis) was also observed in approximately 35% of all observed megakaryocytes (Figure 1). Bone marrow biopsy showed hyper cellularity (100%), with predominance of granulocyte, increased eosinophils and dyspoietic megakaryocytes (Figure 2A). Moreover, an immunostaining performed on bone marrow biopsy using anti-CD117 and -CD34 antibodies did not reveal any significant blastic population (< 2% of all observed medullar cells), nor did the immunotyping (FC500® Series cytometer, Beckman Coulter, USA) using anti-CD34 antibodies on medullar cells (< 2% of all recorded events). Medullar cytogenetic analysis showed a t(5;12)(q33;p13) in 94% of all observed metaphases (Figure 3A and Figure 3B). In addition, cytogenetic and molecular analyses were negative for t(9;22)(q34;q11.2) (FISH based on the use of dual fusion translocation probe and RT-PCR using the LighCycler® LC480, Roche, Switzerland) and JAK2V617F (QX200® droplet digital PCR, Bio-rad, USA). However, an extrachromosomal element was highlighted in 100% of all observed metaphases. Further analysis identified this extra chromosome as constitutional and karyotype could then be defined as 47, XX, t(5;12)(q33;p13), +mar c[26]/47, XX, +mar c[2] (Figure 4A). Based on all results, a diagnostic of myeloid neoplasms with eosinophilia and ETV6-PDGFRB rearrangement was made.
 Figure 1: Pre-treatment blood marrow aspirate showing dyskaryopoiesis A) hypo-lobulated megakaryocytes; B) Micromegakaryocytes; C) Karyorrhexis. May Grünwald Giemsa.View Figure 1
Figure 1: Pre-treatment blood marrow aspirate showing dyskaryopoiesis A) hypo-lobulated megakaryocytes; B) Micromegakaryocytes; C) Karyorrhexis. May Grünwald Giemsa.View Figure 1
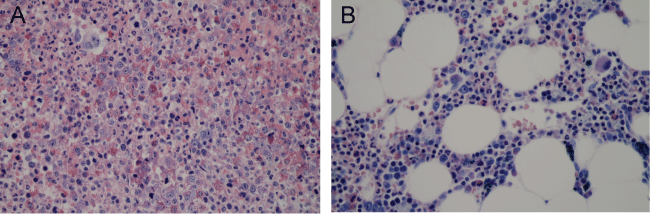 Figure 2: Bone marrow core biopsy. A) (pre-treatment) important cellularity (100%), granulocytic proliferation, increased eosinophils and dysmegakaryopoiesiss; B) (post-treatment) moderate cellularity (50%) without significant dysmyeloplasia, moderate eosinophilia. All three cell lines were represented in physiological proportion. Giemsa stain.View Figure 2
Figure 2: Bone marrow core biopsy. A) (pre-treatment) important cellularity (100%), granulocytic proliferation, increased eosinophils and dysmegakaryopoiesiss; B) (post-treatment) moderate cellularity (50%) without significant dysmyeloplasia, moderate eosinophilia. All three cell lines were represented in physiological proportion. Giemsa stain.View Figure 2
Figure 3: A) (pre-treatment): FISH using a dual-color ETV6 break-apart probe; B) (pre-treatment): FISH using a dual-color PDGFRB break-apart probe; C) (post-treatment): FISH using a dual-color PDGFRB break-apart probe, rearrangements is no longer visualized (sensitivity = 1 to 2% of all analyzed nuclei). FISH was performed using unstimulated cultured bone marrow cells.View Figure 3
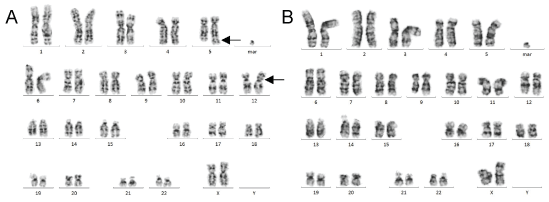 Figure 4: Karyotype of a metaphase from bone marrow aspirate A) (pre-treatment): t(5;12) reciprocal translocation (arrows); B) (post-treatment): absence of t(5;12)(q33;p13) reciprocal translocation. G-banded karyotype, 300 band resolutions, of cultured unstimulated bone marrow cells.View Figure 4
Figure 4: Karyotype of a metaphase from bone marrow aspirate A) (pre-treatment): t(5;12) reciprocal translocation (arrows); B) (post-treatment): absence of t(5;12)(q33;p13) reciprocal translocation. G-banded karyotype, 300 band resolutions, of cultured unstimulated bone marrow cells.View Figure 4
Our patient responded well to a hydroxyurea during the first month with a significant decrease in spleen size and reduction of WBC (including eosinophilia). Treatment with imatinib mesylate was then initiated at a dose of 100 mg/daily, which was progressively increase as described in Table 1 to 400 mg/day without significant adverse effects. Two months after diagnosis and initiation of treatment, peripheral blood count showed disappearance of eosinophilia and normal leukocyte count.
Table 1: Imatinib dosage. View Table 1
As part of follow-up, a bone marrow aspirate and biopsy were performed in September 2016. Bone marrow biopsy showed a cellularity of 50% without obvious histological signs of dysplasia. However, a discrete eosinophilia was observed (Figure 2B). Concerning the bone marrow aspirate, dysmegakaryopoiesis was not visualized anymore and a medullary eosinophilia of 5.4% was assessed (blast = 1.8%). Finally, the t(5;12)(q33;p13) was no longer observed using FISH analysis (Figure 3C). In conclusion, the 54-year-old patient was assumed to be in hematological and cytogenetic remission 11 months after initiation of treatment. Interestingly, imatinib dosage was reduced in April 2017 to 200 mg/daily due to fatigue and diarrhea.
According to the 2008 WHO classification (revised in 2016) of tumours of hematopoietic and lymphoid tissues, the patient falls into the newly proposed category "myeloid neoplasm associated with PDGFRB rearrangement" [1,3]. Prior to this classification update, these neoplasms were often classified as (myelodysplastic)/myeloproliferative disorders such as Chronic Myelomonocytic Leukemia (CMML), Juvenile Myelomonocytic Leukemia (JMML), Atypical Chronic Myeloid Leukemia (ACML), myelodysplasia, eosinophilic leukemia or acute myeloid leukemia [2,4-7].
The incidence of patients with PDGFRB rearrangement is low and accounts for approximately 1.8% of myeloproliferative neoplasms [8]. Depending on case reports and reviews, median age at diagnostic for PDGFRB rearrangement may vary significantly from 42 to 62-years-old. A gender predisposition was highlighted since most of diagnosed cases are male [6,8]. The reasons for this male bias are still unclear and different hypothesis have been made. First, a male-specific mechanism could induce double-stranded breaks in the PDGFRB gene (as an initial event in the development of the leukemia). Second, a female-specific immune or hormonal response against cells expressing ETV6-PDGFRB could block the expansion of this clonal population. Finally, two hits may also be required to cause the development of the disease: a first hit with the translocation and a second one attributed to an unknown X-linked gene [5]. Our case report is interesting as only few reports on female patients with this translocation were described before in the literature.
The fusion protein involving ETV6 and PDGFRB was first described in 1994 in the case of a chronic myelomonocytic leukemia with t(5;12)(q33;p13). Keene, et al. were the first group to link abnormalities of chromosomes 5q and 12p with eosinophilia [5,9]. PDGFRB (anchored in the cellular membrane) belongs to the type III tyrosine kinase receptor family (including other important kinase such as c-KIT, FLT3 or the M-CSF receptor) while ETV6 is a transcription factor belonging to the ETS family members. Within the chimeric protein, the extracellular domain of PDGFRB is replaced by the pointed domain of ETV6, resulting in enforced PDGFRB dimerization and hence constitutive activation of the tyrosine kinase. The subsequent chimeric protein will ultimately stimulate hematopoietic cell proliferation [6,10-12]. Another important aspect of the chimeric protein concerns its physico-chemical properties. When compared to wild-type receptor, PDGFR chimeric protein is more stable and more resistant to degradation due to decreased ubiquitination and proteasomal degradation. This increased stability combined with constitutive tyrosine kinase activity promote cell proliferation [13]. Since the translocation discovery, more than 30 different partners for PDGFRB were observed; some of them are presented in Table 2 [6,14,15].
Table 2: Different PDGFRB gene fusion partners observed in the literature. View Table 2
Using Ba/F3 cells lines or CD34+ hematopoietic stem cells in the absence of growth factor, PDGFRB-ETV6 was shown to activate distinctive signal transduction pathways such as Mitogen-Activated Protein Kinases (MAPK), Phosphatidylinositol-3 Kinase (PI3K) and the transcription factors STAT1, STAT5 and nuclear factor-κB. In addition, experiments based on the use of CD34+ PDGFRB-ETV6+ cells lines showed a strong commitment towards the eosinophilic lineage in liquid cell cultures as it is the case with many patients in vivo [16]. One hypothesis could be the presence of numerous genes of importance in the proliferation and differentiation of eosinophils in the 5q31-5q35 chromosomal region (IL-3, IL-4, IL-5, IL-13 and GM-CSF). Those genes could be dysregulated in the presence of chromosomal rearrangements and lead to a hyper proliferation of the eosinophilic lineage [17].
Clinical findings in patients with PDGFRB rearrangement are heterogeneous. At diagnosis, patients can either be asymptomatic or complain of nonspecific constitutional symptoms such as fatigue or weight loss. Skin involvement was also described [17]. Physical examination reveals mild to massive splenomegaly for most patients while hepatomegaly is less common. CBC counts usually show a hyperleukocytosis together with hypereosinophilia in peripheral blood and/or bone marrow. However, absence of hypereosinophilia can be observed as well. Monocytosis is less common but frequently present. As it was the case here, bone marrow aspirate are generally hypercellular together with a granulocytic hyperplasia. However, cases of dry tap are reported [4,5,18,19].
First-line treatments in patient with ETV6-PDGFRB rearrangements include tyrosine kinase inhibitor (i.e. imatinib mesylate) and are associated with a good clinical, cytological and molecular response [4,7,19]. However, as demonstrated in Table 2 and Table 3, dose of imatinib widely vary between reported patients ranging from 200 mg to 800 mg daily reflecting the uncertainty of physician regarding the optimal dose to be used in this disorder. Resistance to conventional therapy is discussed below.
Table 3: Response to tyrisine kinase (i.e. imatinib mesylate) in patients with myeloid neoplasms associated with ETV6-PDGFRB rearrangement. View Table 3
The follow-up of patients diagnosed with a myeloid neoplasm associated with a t(5;12)(q33;p13) is a crucial issue. In different case reports, similarities were found between the natural course of myeloid neoplasms associated with PDGFRs rearrangements and the three phases of Chronic Myeloid Leukemia (CML): A Chronic Phase (CP), An Accelerated Phase (AP) and A Blast Crisis (BC). Without successful early diagnosis and/or follow-up, the disease can ultimately evolve into BC [20-23]. The evolution to BC is characterized by an accumulation of cytogenetic abnormalities such as trisomy of chromosome 8 [18,23]. Such trisomy was also described in the natural course of CML and associated with poorer prognosis (associated with overexpression of the myc gene located at 8q24) [24]. In addition, due to the accumulation of those cytogenetic abnormalities, patients stop responding to conventional therapies (i.e. tyrosine kinase inhibitors) during AC and BC, hence those patients require other therapeutic approaches. Finally, cases of imatinib resistance were described even in the absence of any cytogenetic evidence of clonal evolution, suggesting the presence of potential unknown mutation which cannot be identified by conventional cytogenetic analysis [18].
Moreover, complications can also be caused by a potential associated chronic eosinophilia which can lead to dysfunction of virtually any organ or organ system including cardiovascular, gastrointestinal, renal, dermatologic, nervous, and upper/lower respiratory systems [18,25]. Concerning the case-report, it could be hypothesized that the heart attack the year before the diagnosis of leukemia could be partly attributed to the hypereosinophilia state. Indeed, eosinophilic count was already at the upper limit of normal range (500/mm3). Unfortunately, this was not investigated at this time. Last, patients can also die from treatment-related complications (e.g. immunocompromised-related or transplant complications) [7].
In case of deep molecular response, trials have shown that CML patients could cease their chemotherapy without risk of relapsing [26]. However, unlike FIP1L1-PDGFRA+ chronic eosinophilic leukemia, it is not yet known if treatment discontinuation could be considered or not for neoplasms associated with PDGFRBs rearrangements. Indeed, imatinib discontinuation in FIP1L1-PDGFRA+ patients has been related to haematological relapses [2,27]. However, insufficient data are currently available to evaluate the potential for imatinib withdrawal for patients diagnosed with neoplasm associated with PDGFRB rearrangements [28].
According to Steer et al., in a compilation of case-reports, the 2-year survival rate of the 19 evaluable patients (19 cases of myeloid leukemia associated with t(5;12)) was only 47% while the median survival was estimated to 20 months. However, some criticism can be brought to those results since they may potentially be biased. Indeed, first, some case reports presented patients who were suspected to be in AP or BC at diagnosis, worsening their survival rate. Second, due to misdiagnosis (e.g. schistosomiasis diagnosed as contact dermatitis instead of eosinophilic infiltration), some patient did not receive appropriate treatment in due time. Finally, imatinib was not always available since some of the case reports were described before the official release of the drug on the market. In addition, a recent retrospective study based on 26 patients (18 ETV6-PDGFRB and 8 others PDGFRB rearrangements) described a 10-year overall survival rate of 90% (95% CI 64-97%) with a median follow up of 10.2 years (1.8-17 years). Once again, due to the rarity of the disease, a publication bias cannot be excluded [28].
In conclusion, we reported here the case of a patient diagnosed with a myeloproliferative neoplasm associated with eosinophilia and rearrangement of PDGFRB with a good response to an imatinib therapy (400 mg/daily). Although imatinib has proven to be an effective therapeutic approach, this mini-review highlighted some difficulties associated with this neoplasm. Firstly, diagnosis is not always an easy task and may be time-consuming, leading to morbidity associated with chronic eosinophilia and inappropriate treatment. Moreover, delayed-treatment can result in disease progression (i.e. accelerated or blast phase). Secondly, natural course of the disease (and other potential unknown mutations) can decrease or stop the therapeutic effectiveness of tyrosine kinase inhibitors. Thirdly, insufficient data are now present in the literature to define the optimal tyrosine kinase inhibitor (i.e. imatinib) dosage and the possibility of treatment withdrawals at hematological and molecular remission state. Finally, further studies will be necessary to better understand the physiological process and the genetics underlying this neoplasm (e.g. male bias, eosinophilic lineage commitment).
None.
None.