40-year-old African man presented with typical anginal pain and cutaneous swelling. Examination revealed that he had corneal arcus, multiple tendon xanthomas and xanthelasmas. The investigation panel showed dyslipidemia with very high total cholesterol of 642.3 mg/dl, very high low-density lipoprotein of 531 mg/dl, low high-density lipoprotein of 36.7 mg/dl and high triglyceride of 248.1 mg/dl and he had a positive stress test. Coronary angiography showed triple vessel disease. The final management plan was a successful coronary artery bypass graft and is currently maintained on lipid lowering drugs, dual antiplatelet and a beta blocker as well as life style modifications.
Hypercholesterolemia is one of risk factors for coronary artery disease. Familial hypercholesterolemia (FH) is a hereditary condition, characterized by elevated low-density lipoprotein cholesterol (LDL-C) right from childhood [1]. FH is often underdiagnosed in many parts of the world, and about less than 10% have been diagnosed with this condition worldwide [2-4]. There are two types of FH; heterozygous FH (HeFH) and homozygous FH (HoFH). The prevalence of HeFH is about 1 in 200-500 while that of HoFH is 1 in 300,000-1,000,000 [1,5]. The prevalence of atherosclerotic cardiovascular disease (ASCVD) was found to be 3 times higher in people with FH than in unaffected people [1,6,7].
Given that FH presents with elevated LDL-C in childhood, it predisposes to early and accelerated ASCVD and xanthomas. Diagnosis is based on three criteria of which Dutch Lipid Network Criteria is widely used. These criteria are mainly based on LDL-C levels, family history, physical findings and premature cardiovascular disease in both patient and relatives available [8,9]. Genetic testing is the gold standard for diagnosis of FH, but not finding a genetic mutation does not exclude the diagnosis especially in the presence of strong phenotype [9]. Cascade screening involves measuring LDL-C levels or genetic testing of relatives to identify those affected so that they can receive treatment and reduce risk of ASCVD [9-11]. Management is directed at lowering LDL-C levels with drug therapy to delay or reduce the development of ASCVD together with life style modifications [1,12,13]. We present a rare case of homozygous FH.
A 40-year-old African businessman with recent diagnosis of hypertension, nondiabetic, nonsmoker and non-alcohol user and with history of premature death of his mother at the age of 49 years. Presented with chest pain for the past 6 months, initial episode was sharp then pressing in nature, and radiating to the back. Chest pain developed while he was exercising and got relieved on resting. This lasted for a short period of time. Second episode of chest pain occurred 4 months later while he was walking and this persisted, worsening on exertion. He reported to have developed cutaneous swellings starting at 7 years of age, initially at the elbow and lower limbs, but progressed to involve the fingers and cubital fossa.
He was seen in a peripheral clinic where he underwent a treadmill stress test of which he stopped at stage 2 due to limiting chest pain and horizontal ST segment depression but no malignant arrhythmias. The attending cardiologist then made a decision to initiate him on optimal medical therapy with Cardiac aspirin 75 mg od, Rosuvastatin 10 mg od, Nitrocontin 2.6 mg od, and Losartan 50 mg od.
About 2-3 months later, he was referred to our facility with persistent anginal symptoms. On assessment, he still reported episodic chest pain especially on exertion but no other associated symptoms.
Young man with palpable peripheral pulses, had corneal arcus, xanthoma at and around the elbow, Achilles tendon, dorsal aspect of metacarpophalangeal and interphalangeal joints, below the knee and lateral aspect of the feet as shown in Figure 1. He was afebrile, no pallor, no jaundice, no edema, and good general condition.
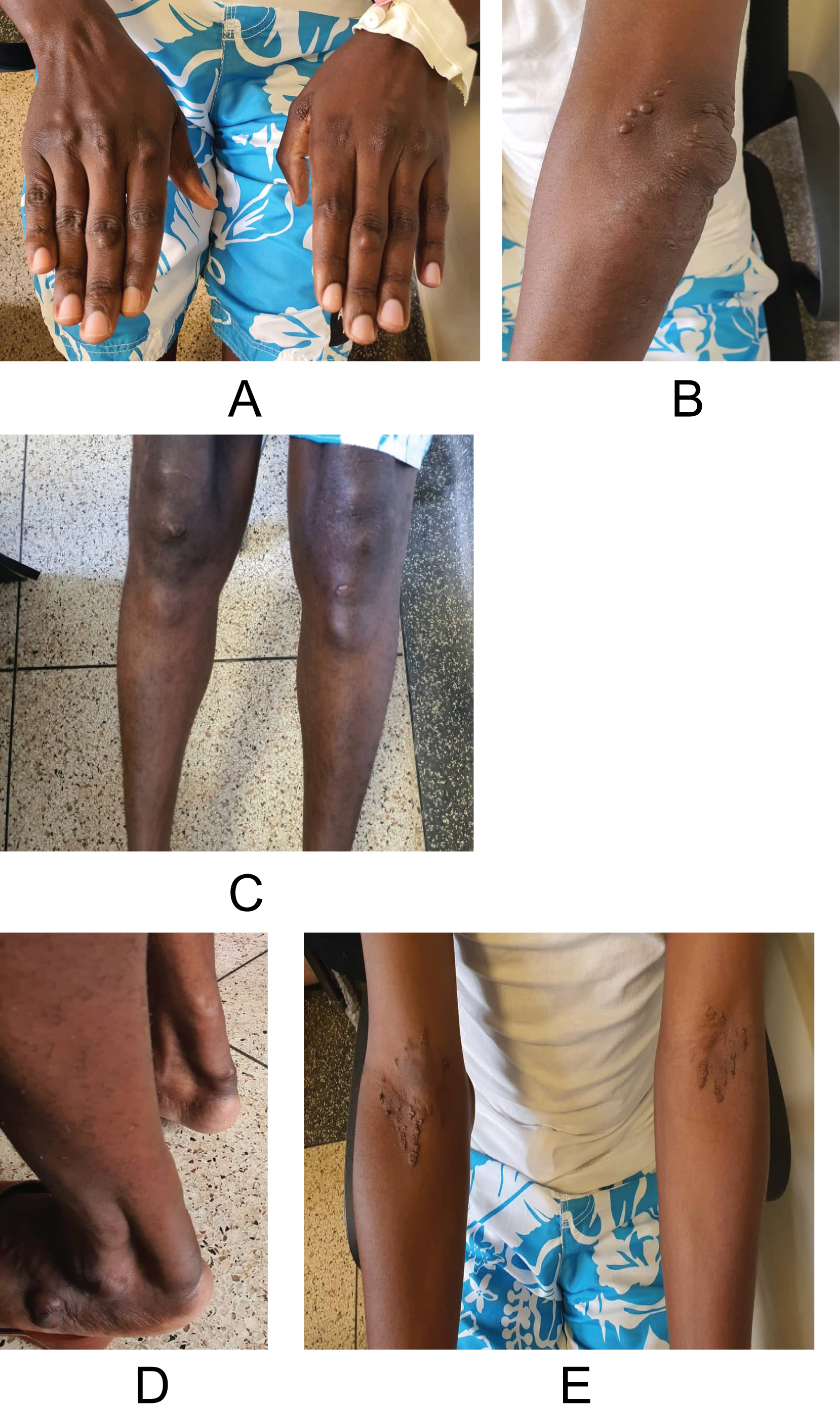 Figure 1: Typical xanthomas in A) dorsal aspect of the fingers; B) at and around the elbow; C) just below the knees; D) Achilles tendon and lateral aspect of feet and E) xanthelasma at the cubital fossa.
View Figure 1
Figure 1: Typical xanthomas in A) dorsal aspect of the fingers; B) at and around the elbow; C) just below the knees; D) Achilles tendon and lateral aspect of feet and E) xanthelasma at the cubital fossa.
View Figure 1
He had a normal heart rate of heart rate of 86/min, blood pressure of the left upper limb154/78 mmHg and right upper limb 148/80 mmHg. Point of maximum impulse was 5 th intercostal space midclavicular line, no raised jugular venous pressure, heart sounds I and II were heard and normal with no added sounds. The respiratory system, abdominal and central nervous system were unremarkable.
Full hemogram, renal function and liver function tests were normal. Hemoglobin A1C was 6.0%, HbsAg was negative, antiHCV antibody negative.
Lipid profile showed very high total cholesterol: 642.3 mg/dl, very high LDL: 531 mg/dl, low HDL: 36.7 mg/dl and high Trig: 248.1 mg/dl.
ECG showed sinus rhythm, no features of ischemia as shown in Figure 2.
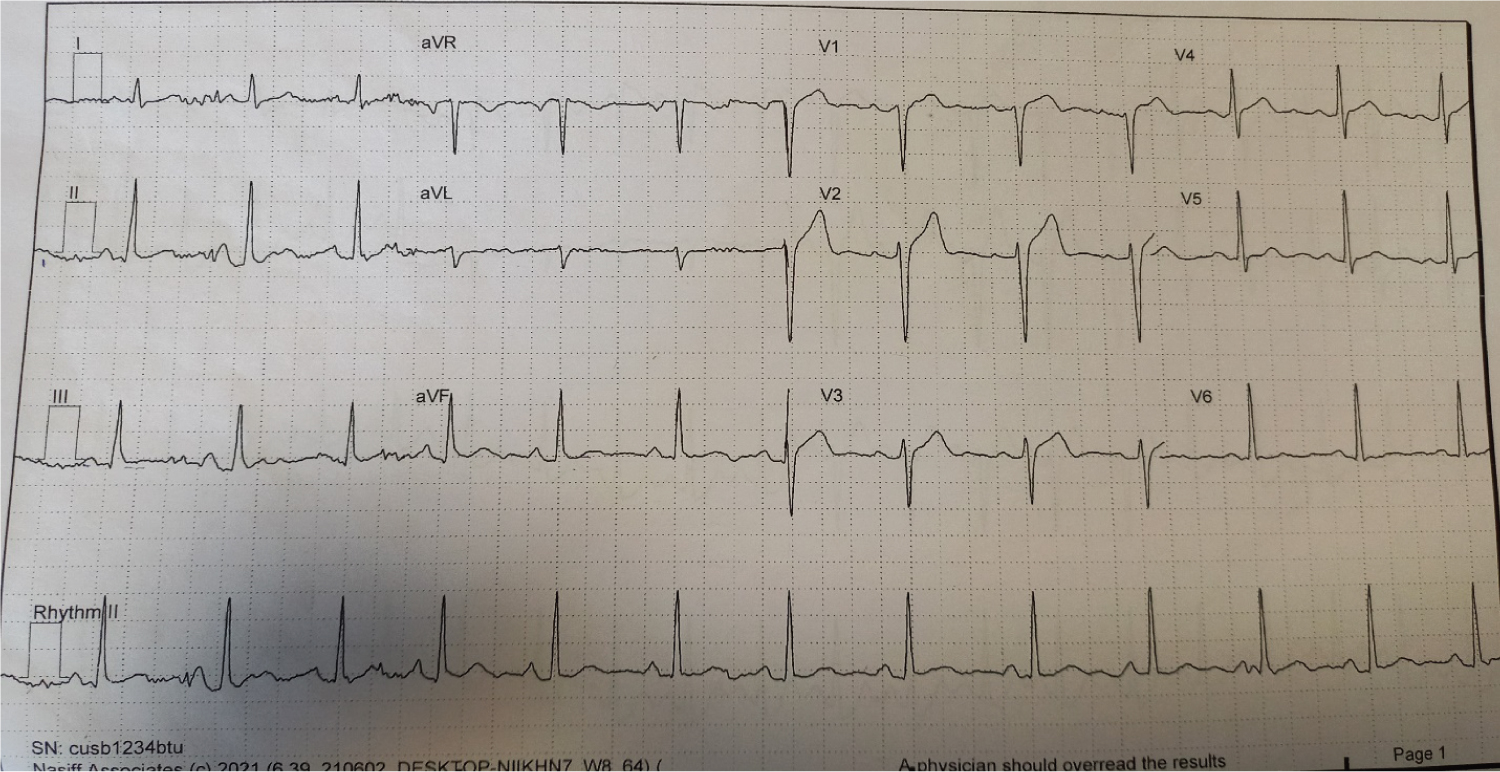 Figure 2: Shows sinus rhythm with heart rate of 78/min, normal QRS axis, normal PR and QTc intervals and normal ST segments (no ischemic features).
View Figure 2
Figure 2: Shows sinus rhythm with heart rate of 78/min, normal QRS axis, normal PR and QTc intervals and normal ST segments (no ischemic features).
View Figure 2
Echocardiogram demonstrated normal biventricular size and systolic function and no evidence of atherosclerotic valve changes or regional wall motion abnormalities. A nodule was taken for histology which showed multiple collections of foamy cells - histiocytes (xanthogranuloma) with no malignant cells seen.
Family members had their lipid profile done; found out that his two brothers and a sister had high LDL-C above 100 mg/dl, however, the father and patient’s daughter of 10 years had normal profile.
He had coronary angiogram which showed severe triple vessel CAD; 80-90% stenotic lesion in proximal left anterior descending artery (LAD), proximal-mid Left circumflex (LCx) 80-90% bifurcation lesion with obtuse marginal 1 and ostio-proximal right coronary artery (RCA) 90% stenosis as shown in Figure 3.
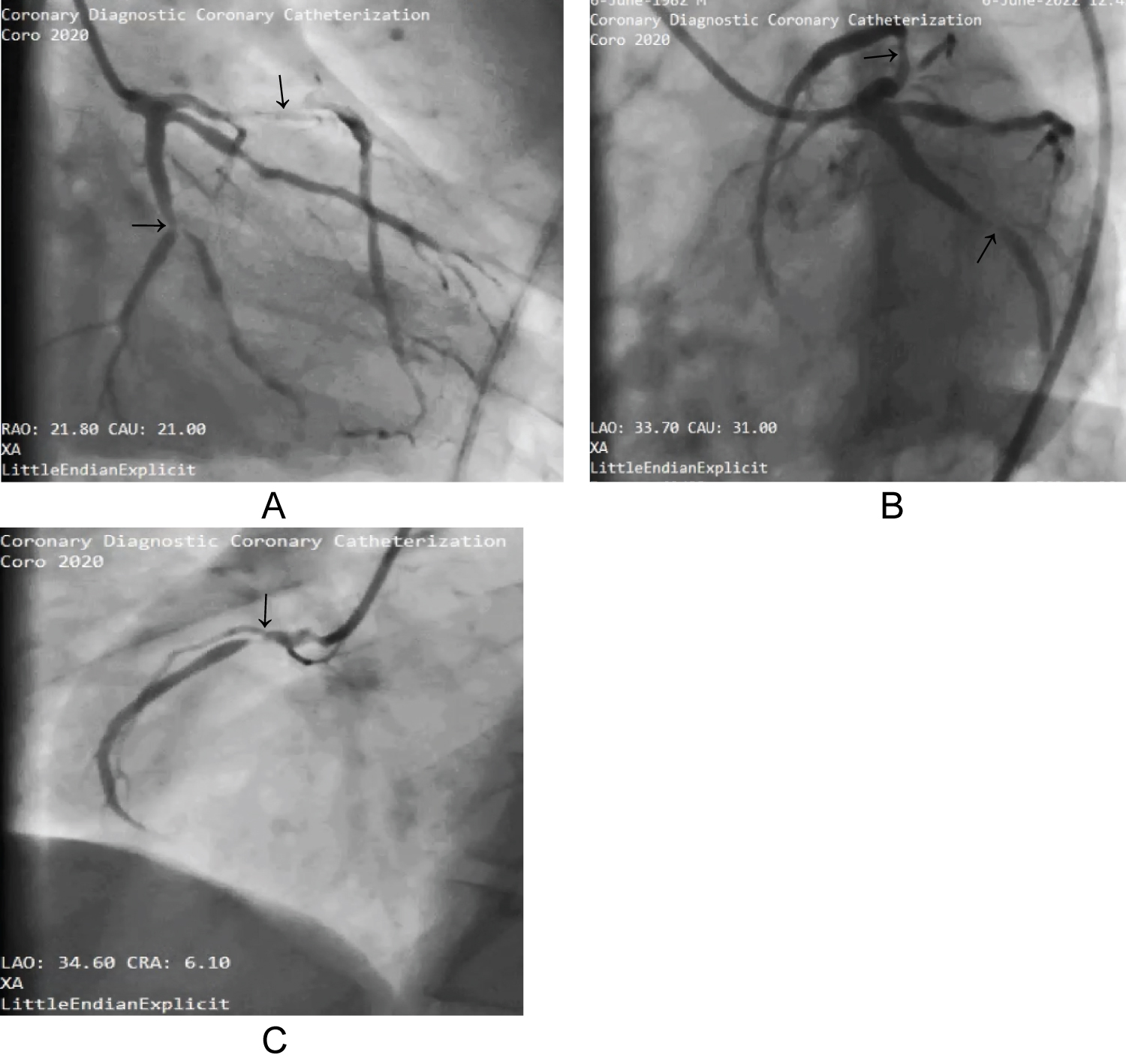 Figure 3: Coronary angiogram A and B shows stenotic lesions in proximal LAD and LCx, stenotic lesion in ostial-proximal RCA in C.
View Figure 3
Figure 3: Coronary angiogram A and B shows stenotic lesions in proximal LAD and LCx, stenotic lesion in ostial-proximal RCA in C.
View Figure 3
Final diagnosis Familial hypercholesterolemia with severe triple vessel Coronary artery disease was made. He was initiated on optimal medical therapy; Rosuvastatin 40 mg od, Ezetimibe 10 mg od, Ecorin 75 mg od, Nebivolol-H 5/12.5 mg od, Telmisartan H 40/12.5 mg od and Nitrocontin 2.6 mg od plus life style modifications.
He was then referred for Coronary Artery Bypass Graft (CABG) which was done successfully with 4 grafts, that is; left internal mammary artery to left anterior descending artery, right internal mammary artery to right coronary artery, left radial artery to - obtuse marginal 1 and obtuse marginal 2 arteries.
He is symptom free lately, we noted slight reduction in xanthoma sizes and lipid profile at 6 months follow up improved to total cholesterol: 228.15 mg/dl, LDL: 189.5 mg/dl, HDL: 38.67 mg/dl, though with an increase of Trig: 522.6 mg/dl.
His current drugs are Clopidogrel/Aspirin 75/75 mg od, Metoprolol succinate 25 mg od, Rosuvastatin 40 mg od and Ezetimibe 10 mg od. The plan is to monitor lipid profile every 6-12 months interval. Addition of PCSK9 inhibitor would be helpful in further reducing the LDL-C, however, they are costly and unavailable here in our country.
Familial hypercholesterolemia (FH) is said to affect about 34 million individual worldwide with the worldwide prevalence varying according to ethnicity [14,15]. We have not come across studies to assess regional and country prevalence of FH but with high index of suspicious, this condition can be easily diagnosed.
FH is an autosomal dominant disorder in which there are defective mutations in the genes that encode for proteins for low-LDL-C receptors [1,9]. The other mutations occur in genes for other molecules that interact with the LDL receptors during LDL-C metabolism; like apolipoprotein B (APOB) dysfunction and proprotein convertase subtilisin-kexin type 9 (PCSK9) gains of function mutation. Because of these defects, there is elevated LDL-C levels starting in childhood [1,6,9]. Other causes of altered lipid metabolism like hypothyroidism, diabetes mellitus, liver disease, nephrotic syndrome and drugs are referred to as secondary causes and should be investigated for. The two variants are homozygous (HoFH) and heterozygous forms (HeFH) of FH.
In HoFH, early exposure to persistently elevated LDL-C leads to accelerated development of ASCVD in childhood and carries a worse prognosis as compared to HeFH [9].
FH manifests as deposits in multiple vascular beds and vascular wall as atheromas and extravascular in the form of xanthomas and xanthelasmas. The three most commonly used criteria to diagnose FH are; Dutch Lipid Network Clinic criteria, the United States (US) Make Early Diagnosis to Prevent Early Death (MEDPED), and Simon Broome Register in the UK. According to the Dutch Lipid Clinic Network Criteria which is widely used, it’s based on LDL-C levels, physical findings, premature cardiovascular disease in relatives and positive genetic testing if available and a definitive diagnosis of FH requires a score of > 8 [8,9].
We present our index case of FH, he had hypercholesterolemia with total elevated cholesterol of 642.3 mg/dl, elevated LDL: 531 mg/dl, low HDL: 36.7 mg/dl, elevated Trig: 248.1 mg/dl. The elevated LDL-C occurs as a result of mutations in genes that encode for proteins involving metabolism of LDL-C, apolipoprotein B and proprotein convertase subtilisin-kexin type 9. Elevated LDL-C starts very early in life, gets deposited in vessel walls, tendons and other tissues hence presenting with xanthomas, xanthelasma and corneal arcus among others. Our patient had corneal arcus, xanthelasma on cubital fossa and xanthomas at and around the elbow, just below the knees, Achilles tendon, dorsal aspect of metacarpophalangeal and interphalangeal joints, and lateral aspect of feet. He had history of premature loss of his mother and exertional chest pain for 6 months.
Genetic testing is the gold standard to diagnose FH, however, we are unable to do it in our setting. In the absence of genetic testing, some diagnostic criteria have been validated and can be used [12]. Using the Dutch Lipid Clinic Network criteria; a definitive FH diagnosis requires > 8 points, a probable FH diagnosis requires 6-8 points and a possible FH diagnosis 3-5 points. Our patient scored 16 points; that is he had premature coronary artery disease which scores 2 points, tendinous xanthomas scoring 6 points and LDL-C > 330 mg/dl scoring 8 points, which confirmed him as definite FH [1,16]. From cascade screening, three of his siblings had elevated LDL-C which is in support of genetic predisposition.
Furthermore, we refer to the case as a homozygous variant of FH in view of the elevated LDL-C and presence of xanthomas before of 10 years of age according to the diagnostic criteria [6]. Genetic testing in HoFH helps to identify those with null mutation in LDL-C receptors which has implications in the treatment [6,12].
This case is of interest because of its an unusual occurrence with prevalence of less than one per million in general population [16].
He had an angiography which revealed triple vessel coronary artery disease which is in keeping with the chest pain that he presented with.
Management of FH is aimed at lowering the LDL-C levels with life style modifications, pharmacotherapy, surgery or lipid apharesis [6,13].
This case being a HoFH variant, usually have minimal response to drug therapy because of the null mutation in LDL-C receptors [6,12]. He however, received high dose tolerable statin with a recommended LDL-C target of less than 55 mg/dl or 50% reduction from baseline [6,13,17]. If the LDL-C target is not achieved, addition of Ezetimibe is recommended, which he also received.
A PCSK 9 inhibitor can be added on the above combination or in cases of failure to tolerate statin, in combination with Ezetimibe. It’s reported that up to 50% require a second line therapy and 20% require PCSK9 inhibitor [13,17,18].
Other drug therapies like Lomitapide, Mipomersen and Bempedoic acid have come on board for those who fail to achieve therapeutic targets [6,13,17,18]. LDL-C apharesis can be combined with pharmacology to facilitate LDL-C reduction by 60% especially in cases with persistently elevated LDL-C, however, its expensive and not readily available [6,19]. Liver transplant and gene therapy have been suggested but still far from reach in our setting [6,20].
Familial hypercholesterolemia is a rare condition and often underdiagnosed. It should be highly suspected in young adult patients who have severely elevated LDL-C above 190 mg/dl, cutaneous manifestations (xanthomas and xanthelasma) with strong family history of premature coronary artery disease. Optimal management aimed at reducing LDL-C levels is key to reduce cardiovascular morbidity and mortality.
We would like to acknowledge all the staff of Coronary Care Unit, all the Faculty for their tireless effort and support and the patient.
Authors declare no conflict of interest.
Uganda Heart Institute.
Justine Namukasa wrote the manuscript, Justine Namukasa, Happy Lilian Mbabazi, Daniel Iraguha conducted patient interview, all authors contributed to interpretation of the case, and all authors reviewed the final manuscript.