High density lipoprotein cholesterol (HDL) has traditionally been considered athero-protective and has been associated with a reduced risk of atherosclerotic cardiovascular disease (ASCVD). This has led to an approach of increasing HDL levels in high risk patients. However, due the complexity of the HDL particle and its diverse role in inflammation and immunity, this simple approach may be detrimental to cardiovascular health. Recent analysis of large population cohorts has established a U-shaped association between HDL and adverse events, further complicating our understanding of the clinical significance of HDL.
In this manuscript, we present two cases of patients with high levels of HDL and atherosclerotic vascular disease. HDL function is discussed and some recommendations as to how physicians should interpret and manage lipid abnormalities in the setting of a raised HDL. We will finish by discussing future directions of research in this undoubtedly complex area.
High density lipoprotein cholesterol (HDL) has historically been considered as "good cholesterol' and has been associated with a reduced risk of atherosclerotic cardiovascular disease (ASCVD) [1]. The Framing-ham study identified a strong positive association between low HDL levels and coronary heart disease [2]. This led to widespread belief that HDL was a modifiable risk factor for ASCVD and thus treatments were developed to increase HDL levels to obtain cardio-protection [3]. Understanding HDL's role in ASCVD is however more perplexing than initially expected. HDL is a complex, heterogenous molecule, with a varied proteome [4]. A large number of proteins involved in inflammation, complement regulation, and innate immunity are physiologically bound to HDL [5]. These reinforce the concept that HDL evolved as a com-ponent of the innate immune system and is reflected in its pleiotropic functions in the body [6]. With its constant remodeling via enzymes and tissues, various subclasses, structural diversity, interactions, and functionality, HDL has a larger role in health and disease [7]. This inverse correlation between raised lev-els of HDL and ASCVD is not necessarily causal [8] and numerous studies have failed to show reduced ASCVD risk with elevation of HDL [9-11]. Contemporary data has highlighted that higher levels of HDL may be detrimental to cardio-vascular health, demonstrating a unique U-shape association between raised HDL and ASCVD risk [12] in men. This paradoxical rise in ASCVD risk is likely driven by several rea-sons including genetic mutation, the concept of HDL dysfunction and inflammation. This had led to an increasing interest in quantifying HDL function and though the reverse cholesterol transport process (RCT) has been given primacy with regard to the proposed mechanistic relationship between HDL and ASCVD, HDL also demonstrates anti-inflammatory, anti-apoptotic, anti-oxidative and vaso-protective properties [13-16].
Our first case is a 54-year-old female who was referred from our colleagues in age related healthcare. She had presented to their service with non-specific neurological symptoms and was subsequently diagnosed with a seizure disorder. MRI of her brain had demonstrated some non-specific white matter foci which were felt to be most likely microvascular in origin. Her lipid profile was deranged with a total cholesterol of 7.1 mmol/L (Normal: < 5.0), Triglycerides of 0.8 mmol/L (Normal < 1.7 mmol/L), low density lipopro-tein (LDL) of 3.7 mmol/L (Normal < 3.0) and high density lipoprotein (HDL) of 3.0 mmol/L (Normal > 1.2). Her referring physician had noted that she had encountered side effects with several different statins and therefore referred this lady to our clinic for consideration of PCSK9 inhibitor therapy. The referral letter specifically mentioned that she had a 'reassuringly high HDL'. Her background medical history in-cluded vitamin B12 deficiency, hypothyroidism secondary to a partial thyroidectomy in 1972, Epilepsy and she was an ex-smoker with a 15 pack year history. Her current medications included: Levothyroxine 125 mcg once daily, levetiracetam 500 mg once daily, aspirin 75 mg once daily and hydroxycobalamin 1000 mcg/ml every three months. We performed a comprehensive biochemical risk marker screen. This revealed a glycosylated hemoglobin of 38 mmol/L (Normal < 42 mmol/L0, c-reactive protein (CRP) of 1.4 (Normal < 5), homocysteine of 9.4 umol/L (Normal = 0-15), Lipoprotein (a) of 482.9 nmol/L (Normal < 72), TSH of 0.16 mU/L (0.3-4.2) and Free T4 of 18 pmol/L (12-22). As mentioned previously, her MRI Brain was suggestive of microvascular disease and her carotid doppler ultrasound (CDUS) revealed mixed type 3 and 4 plaques bilaterally. On review of her "statin intolerance", we discovered that her lipid profile was described as 'not controlled' on atorvastatin 40 mg once daily in 2007. In 2009 she switched to rosu-vastatin but experienced agitation and discontinued therapy. She then trialled simvastatin and experienced muscular stiffness, predominantly in her hands. We started this lady on atorvastatin 20 mg once daily with co-enzyme q10 supplementation. Lercanidipine 10 mg once daily was also started as she was noted to be hypertensive on 24 hour blood pressure monitor. Our patient experienced no issues on statin therapy and her LDL improved to 1.9 mmol/L (Baseline: 3.7 mmol/L), with an increase in HDL to 3.7 mmol/L (Base-line: 3.0 mmol/L).
Our second case is a 57-year-old female who was referred to our risk factor clinic with "a total cholesterol level of 7.6 mmol/L despite lifestyle measures". Her baseline lipid profile showed a total cholesterol of 7.6 mmol/L. A triglyceride level of 0.7 mmol/L, LDL of 4.3 mmol/L and HDL of 3.01 mmol/L. She had no medical history of note, was an ex-smoker with a 5 pack year history. She drank 4 units of alcohol per week and was active, exercising five times per week. Her BMI was normal at 21 and she was normotensive in clinic (119/69 mmHg). Her family history was notable for a father who was diagnosed with angina in his thirties and who underwent coronary artery bypass grafting in his sixties. Of her ten siblings, three were currently taking statin therapy although none had suffered from a myocardial infarction or stroke. She was taking no medications. Additional screening blood-work revealed normal HbA1c, homocysteine and lipoprotein (a) levels. Carotid Doppler ultrasound revealed soft plaque at the level of the carotid bulb bilaterally. We started this lady on atorvastatin 20 mg once daily and continue to follow her up in our clinic.
These two cases demonstrate patients who have developed atherosclerotic disease, despite a supposedly 'protective level' of HDL. They also present with elevated LDL levels. This raises questions regarding our interpretation and management of concurrent lipid abnormalities in the context of a raised HDL level. Ob-servational studies have demonstrated that patients with a lower level of HDL have a higher risk of cardi-ovascular disease [17]. The Framingham cohort also supported this. The primary hypothesis behind this relationship has been the role of HDL in reverse cholesterol transport and as a result, low HDL is now uti-lized as an important component of contemporary cardiovascular risk estimation [18]. Contemporary data has established a U-shaped association between HDL and adverse events in numerous population studies, further complicating our understanding of the clinical significance of HDL [19]. This is further empha-sized by the prospective study of almost two million US veterans, where a U-shaped relationship between HDL level and all-cause mortality was demonstrated [20]. Likewise, a Canadian study highlighted that people with a HDL level > 90 mg/dL were associated with high risk of all-cause mortality [21]. In addi-tion, HDL levels 3.5 mmol/L and < 1.0 mmol/L were associated with high all-cause mortality and high car-diovascular mortality in two prospective Danish cohorts [22]. According to the observations in the above studies, the twopatients described in our study would be categorized as "high risk" due to their HDL con-centration.
More recent analysis has suggested that this relationship may be modulated by other components of the lipid panel. Some studies have suggested that low HDL is not a truly independent risk factor for ASCVD in patients with normal levels of triglyceride and LDL [23]. The authors propose that while high HDL is pro-tective against ASCVD overall, it must be interpreted in the context of concurrent abnormalities in tri-glyceride or LDL levels. Before considering HDL function, it is important to initially convey that High HDL levels do not indicate any reduced risk and should not let physicians discount elevated LDL levels. As observed in the above cases with levels of 3.7 mmol/L and 4.3 mmol/L respectively, the LDL are high, despite the high the HDL. In this regard it would also be important to abandon ratios. As mentioned earli-er, this has important implications as several trials have failed to demonstrate a therapeutic benefit from HDL raising therapies [3-5]. We will discuss these trials in more detail later in the article.
Further evidence challenging the causal relationship between HDL and ASCVD comes from studies show-ing that in some circumstances, high levels of HDL may be associated with an increased risk of ASCVD. Genetic analyses of over 300,000 people confirmed that a genetic variant, called SCARB1 P376L, was associated with elevated HDL levels [24]. The researchers found that people with the variant had unusual-ly high levels of large HDL particles in their blood. To see whether SCARB1 P376L was associated with heart disease, the team acquired data from nearly 50,000 people with coronary heart disease and about 88,000 controls. They found that those with the variant had a significantly higher risk of heart disease. SCARB1 codes for the major HDL receptor on liver cells, scavenger receptor class BI. In mice, genetic manipulations of this gene had effects opposite from those expected if HDL were protective. Over-expression of the gene lowered HDL levels but reduced atherosclerosis. Deletion raised HDL levels but an increased rate of atherosclerosis. Similarly, Apolipoprotein A-1 Milano is a naturally occurring mutated variant of the apolipoprotein A1 protein found in human HDL. ApoA1 Milano is associated with a signifi-cantly reduced incidence of cardiovascular disease, even though it causes a reduction in HDL levels and an increase in triglyceride levels [25]. HDL is a complex particle that is subjected to dynamic remodeling through its interplay with numerous enzymes, tissues, lifestyle and diet, highlighting that the overall un-derstanding of its functions and roles are more intricate than anticipated [7]. These studies provide an intriguing insight into the complexities associated with HDL in vivo. It appears that is not simply a linear relationship with higher HDL levels equating to lower rates of ASCVD.
HDLs as a group of particles can demonstrate intrinsically diverse metabolism and functions depending on their particular proteomic, lipidomic, structural and chemical properties. In addition, HDL carries nu-merous lipids, proteins, enzymes and micro RNAs, which all may play a functional role. The lipidome of HDL has been shown to possess functional properties [26], while the micro RNA profile of HDL can be altered in pathological conditions [27] further emphasizing the dynamic flow of HDL. The lipidome and proteome of HDLs are transformed, with increased triglycerides and decreased phospholipid. Utilization of contemporary methods of classification, such as nuclear magnetic resonance, has increased interest on expounding the complex nature of lipidome, proteome, and structural difference of HDL particles [26,28-30].
This leads us to the concept of 'functional HDL deficiency'. This refers to cases where, despite normal levels of HDL on quantitative measurement, patients may be functionally deficient. This functional defi-ciency may result from HDL losing its function and becoming less athero-protective ('loss of function'), or alternatively, HDL may become dysfunctional, resulting in pro-inflammatory effects ('gain of dysfunc-tion').
Defining functional HDL deficiency or HDL dysfunction has been attempted in several different ways. This can include assessing for oxidative modification of HDL particles and utilizing functional assays. These functional assays can test for, amongst others; macrophage cholesterol efflux and the ability of HDL to inhibit monocyte chemotaxis. Some authors have proposed the concept of an 'inflammatory in-dex' for HDL, which is defined as the relative capacity of HDL to inhibit monocyte chemotaxis induced by oxidized LDL. This monocyte chemotaxis assay has been shown to distinguish between ASCVD and control individuals with higher accuracy than the measurement of HDL levels [31].
Functional assays may not be scalable and feasible at present. An alternative approach may be to assess the HDL proteome in order to identify changes in the protein composition of HDL. These changes may be surrogate markers of altered HDL function. Surrogate markers may include changes in the size/density of HDL particles and the specific proteome present. Identification of these alterations in HDL may confer physicians with a better ability to predict cardiovascular risk. Certain subtypes of HDL have already been identified which seem to predict cardiovascular risk more accurately than HDL alone [32]. We will dis-cuss some of these in the next section.
Apolipoprotein AI (ApoA1) is the major protein component found in HDL. It is considered to play a vital role in the reverse cholesterol transport process as shown in Figure 1. However, it may undergo post-translational modification by myeloperoxidase (MPO), which can impair its function as a cholesterol ac-ceptor. MPO can induce molecular alterations in ApoA1 including nitration and chlorination of specific apoAI tyrosine residues. This is associated with specific loss of ABCA1 mediated cholesterol acceptor activity. As a result apoAI cholesterol acceptor activity is inversely correlated with ApoAI nitro- and chloro-tyrosine levels. Unsurprisingly, plasma ApoAI nitro- and chloro-tyrosine levels are higher in ASCVD patients than in controls and plasma MPO levels have been positively associated with ASCVD and the risk of MACE [33,34]. Some authors have proposed that levels of nitro- and chloro-tyrosine in ApoA1 may serve as useful markers of MPO activity in vivo. This oxidative modification may be associ-ated with several conditions; chronic kidney disease, metabolic syndrome and type 2 diabetes mellitus. There have been other modes of oxidative modifications of apoA-I beyond nitration or chlorination doc-umented. In a study by Hazen, et al., Tryptophan modification of apoAI was primarily responsible for the myeloperoxidase-mediated loss of the cholesterol acceptor activity of apoAI [35].
Sphingosine-1-Phosphate (S1P) is another proposed mechanistic cause for HDL dysfunction. It is a lipid mediator found in the erythrocytes, platelets and endothelial cells, and binds to the lysophospholipid re-ceptor family. Only S1P-bound HDL (60% of total plasma S1P) seems to be active, whereas albumin-bound S1P (40% of plasma S1P) acts as a reservoir. One study discovered that HDL oxidation resulted in a 44% loss of HDL-S1P compared to native HDL [36]. Western blot analysis of apolipoprotein AI (ApoAI) as the major HDL apolipoprotein and apolipoprotein M (ApoM) as an important S1P binding partner in HDL showed that both proteins were altered by oxidation: ApoM was virtually absent in oxidized HDL, whereas ApoAI appeared as several immunoreactive bands. It has been observed that healthy HDL and ASCVD HDL with the same S1P content are equally efficient. The authors proposed that HDL signaling was mainly dependent on their ability to carry, retain, and present S1P. Interestingly, this group was sub-sequently able to artificially increase the S1P content of HDL, thereby increasing its function. This may represent a potential future therapeutic strategy to restore HDL function.
Serum Amyloid A (SAA) is an acute phase HDL-associated apolipoprotein produced primarily by hepato-cytes [37]. SAA is secreted during the acute phase of inflammation and has several immunomodulatory roles. These include neutrophil activation and cytokine induction [38]. This may play a key role in local and systemic inflammation and free fatty acid production, promoting ASCVD [39]. SAA also enables the production of reactive oxygen species (ROS) through its chemotactic ligand properties and indirect affect on the potent chemotactic factor, fMLP [40]. This may similarly promote the establishment and advance-ment of ASCVD [41]. A review by Egom, et al. highlights the literature giving weight to the theory that the S1P/SAA Index may affect the HDL athero-protective properties and thus indicate a potential target for therapeutic interventions. Increased SAA was determined to be highly associated with increased all-cause and cardiovascular mortality [42].
Many studies have demonstrated raised serum SAA levels in patients with ASCVD [43]. It has also been shown to be elevated in high cardiovascular risk populations including smokers and patients with diabetes mellitus and chronic kidney disease [44,45]. Raised serum levels of SAA may inhibit endothelial NO pro-duction, increase production of ROS and impair reverse cholesterol transport [43], Patients with ASCVD have also been demonstrated to express considerably higher levels of SAA in HDL particles in compari-son to healthy subjects [46]. This accumulation of SAA into HDL particles may disrupt their anti-atherothrombotic mechanisms [47,48], leading to HDL dysfunction [49,50].
Based on the data discussed above, SAA may be considered the 'inflammatory marker' and S1P the 'athero-thrombotic marker' with regard to HDL function. Evaluating both markers concurrently may pro-vide physicians with a comprehensive overview of HDL function.
Apolipoprotein C-III (ApoC-III) is a pro-inflammatory protein that resides on the surface of very-low-density lipoprotein (VLDL), LDL, and HDL particles. Presence of apoC-III in HDL sub-fractions from plasma may identify participants with an increased risk of future CHD [51]. ApoC-III inhibits lipoprotein lipase and HDL particles bearing ApoC-III are associated with an increased rather than a decreased risk of ASCVD. It has been postulated that the presence/absence of apoC-III in HDL sub-fractions from plasma could be used to stratify an individual's future risk of ASCVD. One study [52] reported that the relative risk of CHD per standard deviation of HDL without ApoC-III was 0.66 (95% CI 0.53-0.93), whereas it was 1.18 (95% CI 1.03-1.34) for HDL with ApoC-III; these findings indicate that HDL subtypes without ApoC-III were inversely associated with the risk of CHD, whereas HDL subtypes with ApoC-III showed a direct associationwith the risk of CHD. These data suggest that ApoC-III interferes with the atheroprotec-tive function of HDL. With regards to the role of lipases, deLemos, et al. undertook a genome-wide asso-ciation study that described several variations of the endothelial lipase gene that were exclusively associ-ated with HDL [53]. One specific loss of function of single nucleotide polymorphism (Asn396Ser) was associated with a substantialrise in HDL and reduced endothelial lipase activity in vitro [54] potentially correlating the role of lipases with the U-shaped curve discussed earlier.
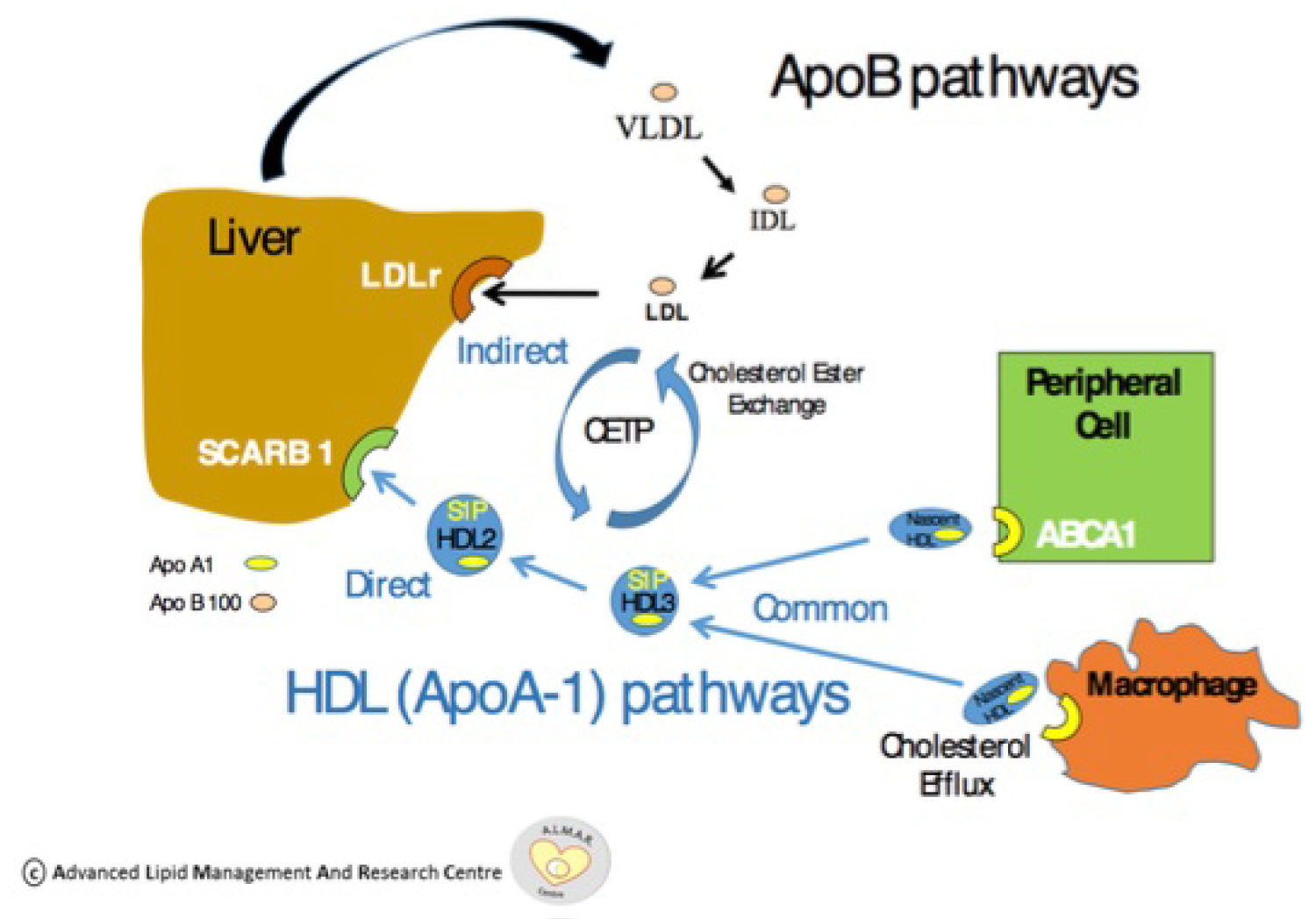 Figure 1: ApoA1 pathway and effect on reverse cholesterol transport. Reproduced courtesy of Prof. Vincent Maher of the Advanced Lipid Management and Research (ALMAR) Centre at Tallaght University Hospital
View Figure 1
Figure 1: ApoA1 pathway and effect on reverse cholesterol transport. Reproduced courtesy of Prof. Vincent Maher of the Advanced Lipid Management and Research (ALMAR) Centre at Tallaght University Hospital
View Figure 1
This paradigm shift to a qualitative rather than quantitative assessment of HDL might explain the failure of several HDL raising therapies to demonstrate a reduction in cardiovascular events. This has been par-ticularly noted with the CETP inhibitors, torcetrapib and dalcetrapib. In the ILLUMINATE study [10], Torcetrapib led to an increase in all-cause mortality and cardiovascular events, although it was able to increase HDL by 72% and decrease LDL by 25%. It should be noted that unfortunately, torcetrapib also increases blood pressure and increases aldosterone as an off-target, non–mechanism-based effect. In the dal-OUTCOMES [11] study, dalcetrapib increased HDL cholesterol levels but did not reduce the risk of recurrent cardiovascular events in patients who had had a recent acute coronary syndrome. These studies provide further insight into HDL function and indicate that a focus on simply raising HDL levels will not be sufficient to improve cardiovascular outcomes. Potential therapeutic avenues in the future may include oxidation resistant HDL, oxidation resistant APoA1 and artificial elevation of S1P in order to restore HDL function.
As we can see from this article, the relationship between cardiovascular disease and HDL is complex. This complexity reflects the varied proteome and pleiotropic functions associated with HDL in vivo. It appears that quantification of HDL levels is not always sufficient to predict cardiovascular risk. As can be seen in our two cases, patients can still develop cardiovascular disease despite levels of HDL traditionally thought of as athero-protective.
Much progress has been made with regard to assessment of the HDL proteome and HDL function. The hope is that in the future, this information may translate to improved risk stratification and therapeutic strategies in order to reduce the risk of cardiovascular disease. However much work remains in order to further clarify the optimum diagnostic and therapeutic approach.
In the interim period, physicians should remain cognizant that HDL may not always be atheroprotective. As such HDL levels must not be viewed in isolation and should be interpreted in the context of concomi-tant lipid abnormalities in order to determine the optimum therapeutic strategy for an individual patient. It seems that with regard to HDL, quality is more important than quantity.
HDL values need to be interpreted in the context of concomitant lipid abnormalities and the overall risk factor profile of the patient.
High levels of HDL may not always be protective.
In the future, functional testing of HDL may provide further information for physicians to estimate cardi-ovascular risk and decide on optimum treatment strategies. Future therapeutic modalities may aim to re-store and optimize HDL function rather than to simply increase HDL quantity.
This study received no funding.
The authors report no relationships that could be construed as a conflict of interest.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants included in the study.