Systemic polyarteritis nodosa is a vasculitis characterized by inflammation and fibrinoid necrosis of medium and small arteries, with a global incidence of 0.7/100,000 and a prevalence of 6.3/100,000. Its cause is as yet unknown, and it affects both children and adults. The clinical presentation tends to be insidious and vague in its initial stages. Cardiac involvement is one of the signs of severity, with coronary artery impairment and aneurysmal lesions of varying degrees. The consequent heart failure and acute myocardial infarction are complications which increase mortality. Without treatment, five-year survival is 13%. Medical treatment is aimed at halting the inflammatory process and treating the consequences of the dysfunction of various organs and systems. We present a case of an eight-year-old boy with polyarteritis nodosa who developed giant aneurysms of the coronary arteries with severe cardiac dysfunction and multisystem impairment. The proper diagnosis and prompt treatment achieved clinical recovery and early survival.
Polyarteritis nodosa, Coronary artery aneurysms, Multisystemic vasculitis, Necrotizing vasculitis
Polyarteritis nodosa (PAN) was first described in 1852 by the German pathologist from Vienna, Karl von Rokitansky, in a 23-year-old man with a five-day history of fever and diarrhea [1]. It is a systemic necrotizing vasculitis characterized by inflammation and fibrinoid necrosis of medium and small arteries [2]. It has a global incidence of 0.7/100,000 and a prevalence of 6.3/100,000 [3,4]. Its etiology is unknown, but it has been related to various infectious processes. Between 10-50% of adult cases are related to the hepatitis B virus, and some are related to the hepatitis C virus. Case reports in children are scarce [5]. In 2004, Ozen, et al. published a multicenter review with 110 pediatric patients, finding a relationship with hepatitis B and C in only 4.6% of cases [6]. Typically, it presents as a systemic process, characterized by fever and general malaise, asthenia, anorexia, fatigue, and weight loss. Various organs and systems (cardiovascular, renal, gastrointestinal, musculoskeletal, and nervous system, among others) are affected. Local and systemic manifestations depend on the disease severity and organ involvement, and include maculopapular purpuric lesions which may be complicated by necrotic lesions and peripheral skin gangrene, as well as proteinuria and glomerulonephritis which may progress to kidney failure [7]. From a cardiovascular standpoint, PAN may affect the coronary arteries (CAs), with the development of aneurysmal lesions of varying sizes which progress to heart failure (HF), acute myocardial infarction, myocarditis, and conduction disorders, among others [8-10].
This was an eight-year-old male patient who had been hospitalized nine months before due to prolonged fever, maculopapular skin lesions on his upper limbs, myalgia, arthralgias and peripheral edema. He had leukocytosis and elevated C-reactive protein (CRP). His echocardiogram was normal. Kawasaki disease (KD) was suspected, and he received intravenous immunoglobulin in addition to intravenous penicillin for suspected sepsis. His symptoms improved and he was discharged after 30 days. He did not report any monitoring or follow up. He was referred to our cardiovascular center with a two-month history of deteriorating functional class (FC), weight loss, intermittent fever (39 °C), and progressive dyspnea. On physical exam, his heart rate was 138/min, respiratory rate 32/min, blood pressure 100/60 mmHg, temperature 37 °C, weight 26 kg, and height 160 cm, with enlarged neck and inguinal lymph nodes. His heart had a regular rhythm, with low-pitched first and second sounds and no murmurs. A chest X-ray showed cardiomegaly. An electrocardiogram showed sinus tachycardia with no signs of ischemia. An echocardiogram showed a large pericardial effusion, left ventricular (LV) dilation, and severe systolic dysfunction with an ejection fraction (EF) of 33%; the CAs could not be evaluated. A pericardial window was done, extracting 350 mL of clear liquid (transudate) whose culture and immunological studies were negative. The clinical deterioration persisted (hepatomegaly, edema of the lower limbs and systemic arterial hypertension [SAH]), with ongoing leukocytosis and neutrophilia, elevated erythrocyte sedimentation rate and CRP, as well as proteinuria and ascites, and no improvement with medical treatment for HF.
A heart catheterization revealed a giant aneurysm of the left main coronary artery (LCA) and right coronary artery (RCA), with multiple aneurysmal dilations (the aneurysmal rosary sign) (Figure 1 and Video 1). In addition, there were diffuse small vessel lesions on the distal pulmonary artery, and mesenteric and renal arteries (Figure 2 and Video 1). Thus, systemic PAN was diagnosed. Intravenous corticosteroids, cyclophosphamide and platelet antiaggregation (acetylsalicylic acid + clopidogrel) were administered. He was also treated for HF with carvedilol, enalapril, furosemide and spironolactone. Follow up one year later showed a normal-sized LV, 60% EF, and persistent lesions in the CAs. The proximal third of the LCA was 19.2 mm × 18.1 mm, and the RCA was 13.9 mm × 14.2 mm. Angiography of the renal and pulmonary vessels showed that the lesions had resolved (Figure 3). He was in FC I, with no acute ischemic events.
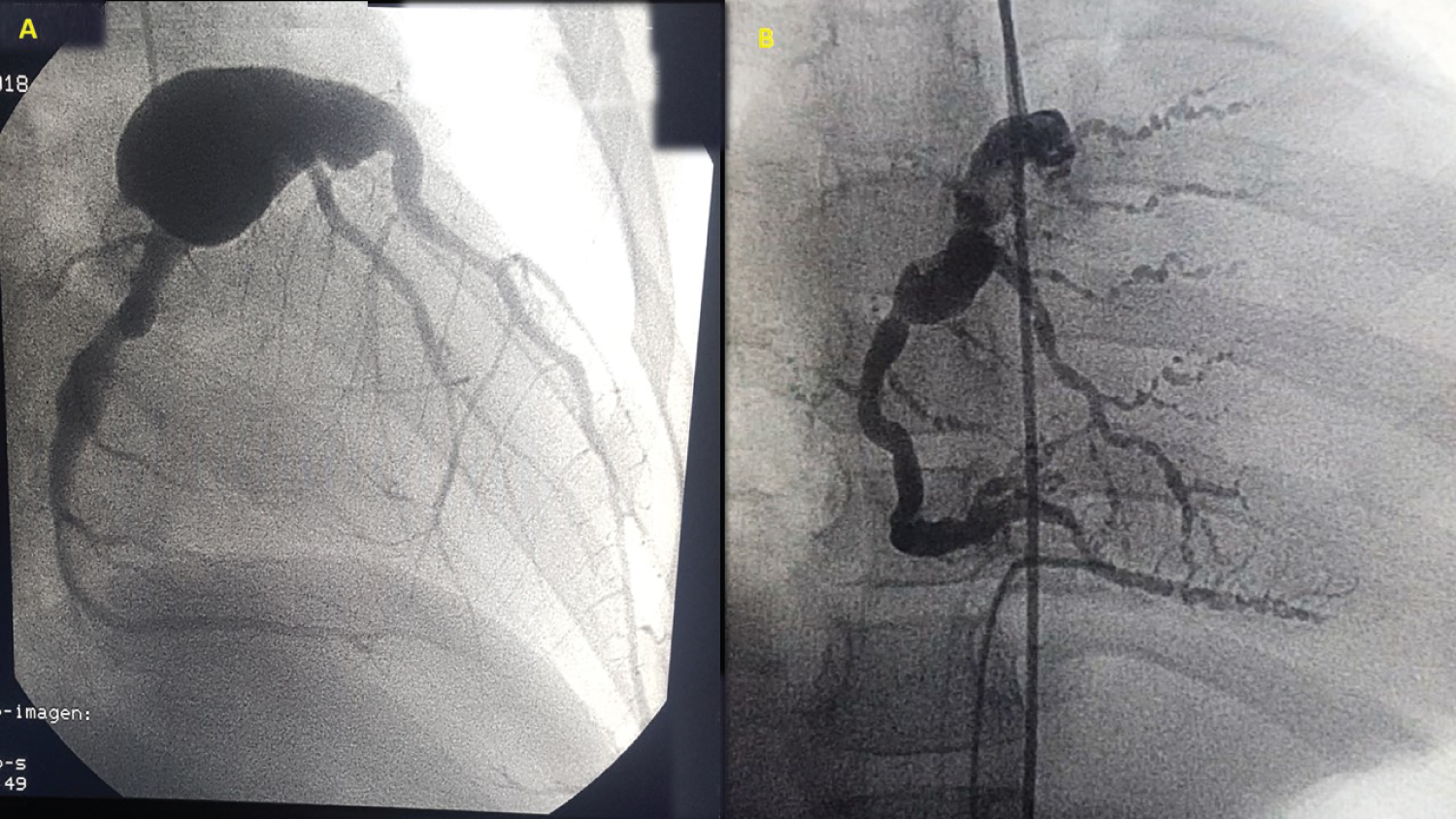 Figure 1: (A) Coronary catheterization showing a giant aneurysm affecting the left main coronary artery; (B) Right coronary artery, with multiple aneurysmal dilations (the aneurysmal rosary sign).
View Figure 1
Figure 1: (A) Coronary catheterization showing a giant aneurysm affecting the left main coronary artery; (B) Right coronary artery, with multiple aneurysmal dilations (the aneurysmal rosary sign).
View Figure 1
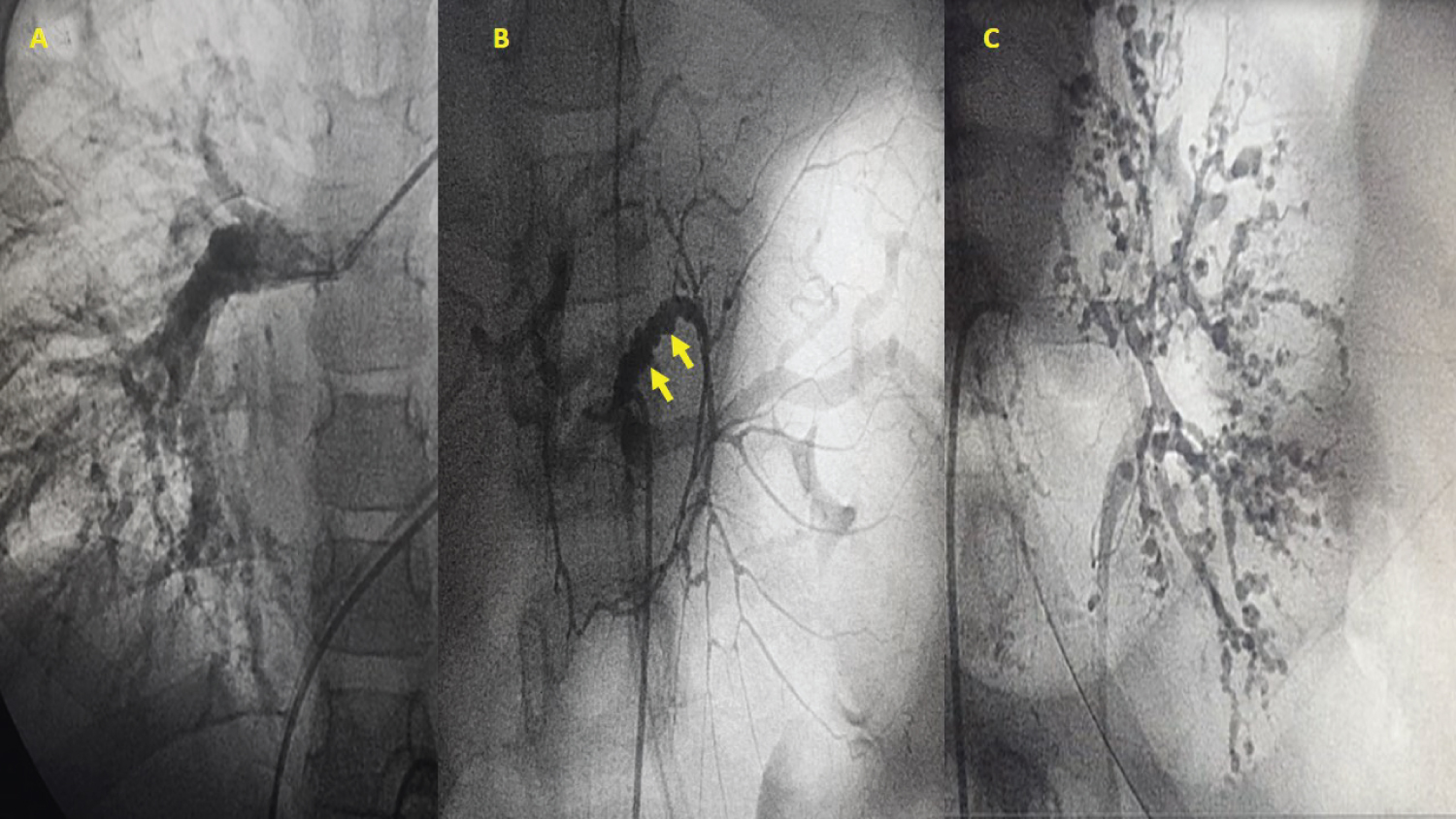 Figure 2: (A) Pulmonary artery angiography showing diffuse small vessel lesions on the distal right pulmonary artery; (B) Catheterization of the abdominal aorta; mesenteric angiography shows multiple saccular vascular lesions (yellow arrows); (C) Left renal catheterization; angiography shows diffuse small vessel lesions.
View Figure 2
Figure 2: (A) Pulmonary artery angiography showing diffuse small vessel lesions on the distal right pulmonary artery; (B) Catheterization of the abdominal aorta; mesenteric angiography shows multiple saccular vascular lesions (yellow arrows); (C) Left renal catheterization; angiography shows diffuse small vessel lesions.
View Figure 2
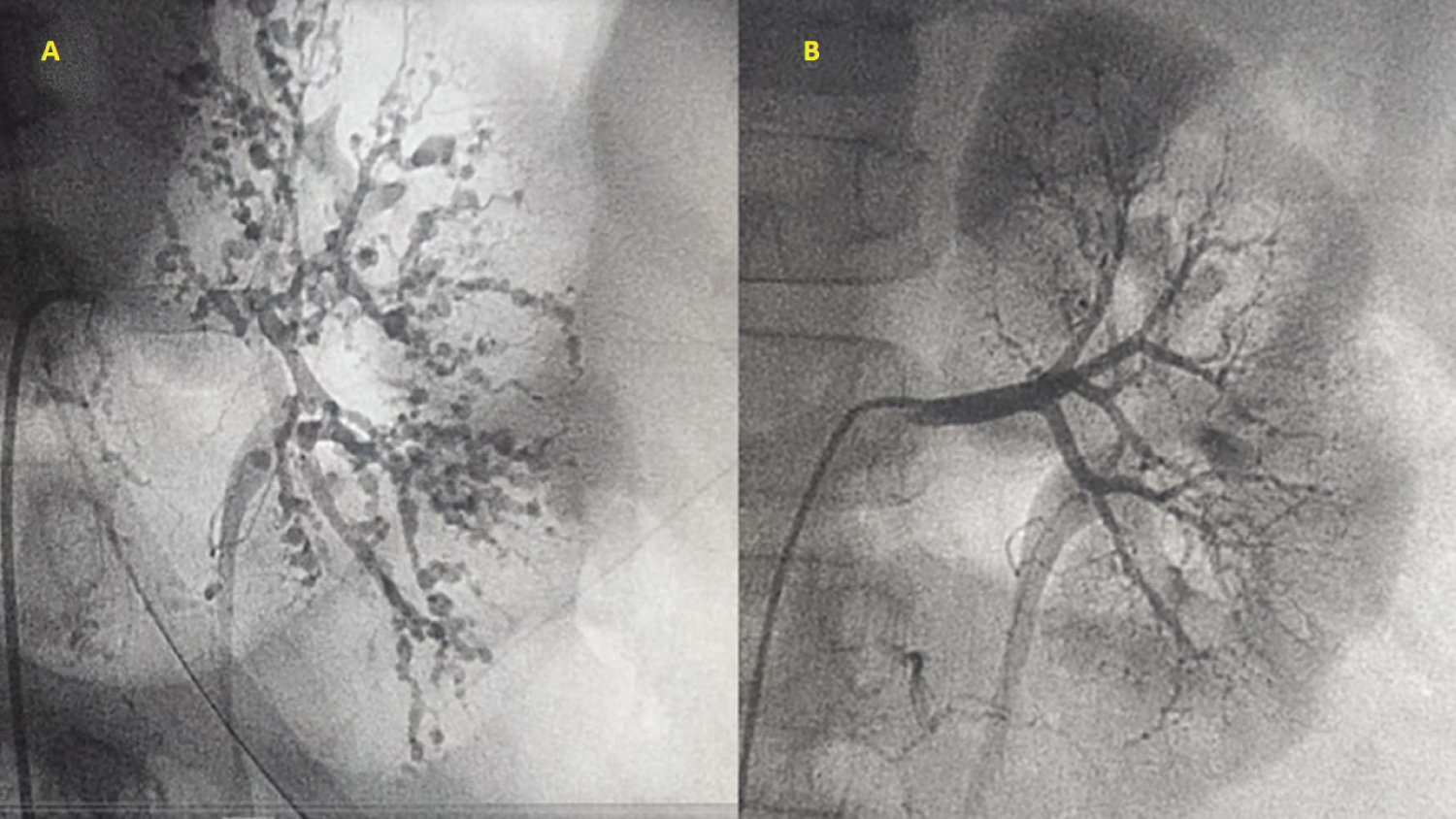 Figure 3: Comparative angiography of the left renal arteries: (A) Initial lesions and (B) After successful treatment and one year of follow up.
View Figure 3
Figure 3: Comparative angiography of the left renal arteries: (A) Initial lesions and (B) After successful treatment and one year of follow up.
View Figure 3
Video 1: In order: Right and left renal catheterization followed by catheterization of the abdominal aorta, mesenteric angiography and, finally, right coronary artery angiography. View Video 1
As a multisystemic vasculitis, the clinical presentation of systemic PAN may range from a self-limited disease with no sequelae to fulminant multisystem failure. Most cases are of the serious type [11]. The classic presentation in childhood is insidious, with symptoms which include SAH in up to 80% of cases, fever, general malaise, fatigue, anorexia, weight loss, myalgia, muscular sensitivity, eye symptoms and testicular pain [12,13]. Medium-vessel vasculitides include PAN, cutaneous polyarteritis and KD. These vasculitides have common symptoms such as persistent fever and constitutional symptoms like weight loss, myalgia and arthralgias. These symptoms are accompanied by purpuric skin lesions and altered lab tests indicative of inflammation such as leukocytosis, eosinophilia, increased erythrocyte sedimentation rate, and elevated CRP, among others. Systemic signs of dysfunction will depend on the organs and systems (cardiovascular, renal, digestive, musculoskeletal) involved. Untreated KD, specifically, may affect the CAs in 15-25% of cases, with the development of aneurysms which may lead to myocardial infarction, sudden death or ischemic heart disease [14]. The patient's first assessment was performed outside of our institution. We postulate that the physicians thought of a possible KD due to the previously described symptoms, and decided to administer intravenous immunoglobulin treatment. This may have caused transient symptom resolution and delayed the PAN diagnosis. However, in our institutional assessment, we no found past history about bilateral conjunctivitis, inflammation of mucous membranes of the mouth and throat (dry, red, cracked lips and a strawberry-red tongue) swelling of lymph nodes in the neck (cervical lymphadenopathy); redness and swelling of the hands and feet, peeling skin on the hands and feet. We propose the hypothesis that, given the findings at our cardiovascular center, PAN may have begun from the onset of symptoms. Although KD has some symptoms in common with PAN, it is clear that its occurrence is uncommon in children over the age of five [15].
Heart involvement in systemic PAN is unusual, and occurs in 5 to 20% of cases, generally in the context of other organ and system involvement. Coronary artery aneurysms occur most often in the RCA, followed by the left anterior descending artery and, finally, the circumflex; they rarely involve both main arteries and their divisions [16,17]. In the presented case, we found a giant LCA aneurysm on the main trunk, associated with an RC aneurysm. This explains the HF which developed prior to being admitted to our institution. Despite the giant size of the LC aneurysm, the CAs could not be assessed in the first echocardiographic study due to the large pericardial effusion and persistent tachycardia which made a detailed evaluation difficult. However, when HF is present, various congenital abnormalities of the coronary circulation must be ruled out (anomalous left coronary artery from the pulmonary artery [ALCAPA]). However, there were no signs of ischemia on the electrocardiogram, which made the diagnosis more difficult. Systemic arterial hypertension is a very common finding, but in our case it was not found initially, due to great hemodynamic instability and signs of imminent cardiac tamponade secondary to the large pericardial effusion (as evidenced by tachycardia and normal arterial pressure). Once the pericardial effusion resolved, SAH was revealed, providing another diagnostic criterion for systemic PAN. In light of the unexplained cause of HF, coronary angiography was key for diagnosing the CA lesions. The characteristic sign of an “aneurysmal rosary” is described in the context of multisystem involvement, and is often seen in the mesenteric arteries in systemic PAN; this was found in the RC (Figure 2). Kawasaki disease, Behçet's disease, Takayasu's arteritis, giant cell arteritis or syphilitic arteritis, and ALCAPA should be ruled out in the differential diagnosis.
Systemic PAN is diagnosed according to the criteria proposed by the European League Against Rheumatism/Pediatric Rheumatology International Trials Organisation/Pediatric Rheumatology European Society (EULAR/PRINTO/PRES), respectively, which identify necrotizing vasculitis when at least one minor criterion is present along with an angiographic anomaly such as an aneurysm, stenosis or occlusion of a medium or small artery The diagnosis of systemic PAN has a sensitivity and specificity of 89.6% and 99.6%, respectively [18]. The case presented met the classification criteria for a diagnosis of systemic PAN (SAH, non-nephrotic proteinuria and myalgias), together with multiple disorders of the coronary and renal vessels.
Currently, the recommended treatment of choice for systemic PAN aims to decrease systemic vascular inflammation. The administration of steroids combined with cyclophosphamide or azathioprine has a greater than 80% effectiveness [19]. Suggested alternatives include methotrexate, mycophenolate mofetil, immunoglobulin, infliximab or rituximab. However, these results are based on studies in adults. In our case, there was a good response to corticosteroids and cyclophosphamide, associated with good HF control. However, after successful treatment, the disease recurs in 10-20% of patients [20]. Likewise, when CA aneurysms are present, platelet antiaggregation is mandatory [21].
Without treatment, the five-year survival is 13%. With optimal treatment, five-year survival increases approximately 80%, with a mortality of approximately 1-4% [22].
Systemic PAN is a rare, progressive disease with an insidious presentation. The prognosis is determined by the involvement of multiple organs and systems. This case highlights a significant involvement of the CAs, developing giant aneurysms and significantly affecting cardiac function. Due to the insidious presentation, the diagnosis tends to be unclear and may be delayed, as in this case. It is important to differentiate PAN from other types of vasculitis, including KD. The diagnostic guidelines proposed by EULAR/PRINTO/PRES must be followed to avoid delayed diagnosis and treatment. This caseteaches us that, in the presence of HF or fever of unknown origin, and once the more common diseases are ruled out, systemic PAN must be suspected. Angiographic diagnosis is definitive for detecting CA lesions, and treatment aimed at stopping the inflammation and avoiding greater consequences should be promptly instated. Between 10-20% of systemic PAN patients relapse; thus, specialized follow up is necessary, including complementary CA imaging, as well as antiplatelet therapy.