The author has just written a book on the evolution of the specialty of Pediatric Cardiology over the last 50 years [1]. The intent of this review is to present a summary of this book. Because of large amount of the material, the review is separated into four parts. In the Parts I and II, percutaneous interventional procedures were discussed. The Part III reviewed electrocardiography, echocardiography, and cardiac catheterization. In this last paper, subjects that were not discussed in the first three reviews will be presented.
In patients with complex heart defects, ventricular septal defect (VSD) is important in maintaining appropriate intra-cardiac shunt; we have recommended that such VSDs are named physiologically advantageous VSD [2]. Spontaneous closure of isolated VSDs was well documented in the 1960s and 1970s [3-8]; yet, closure of physiologically advantageous VSDs was not adequately documented in prior studies [2]. The author initially witnessed spontaneous closure of physiologically advantageous VSDs in two children, one with tricuspid atresia and the other with double outlet right ventricle [2]. This enticed the author to examine this phenomenon further. This issue was thoroughly examined in patients with tricuspid atresia in several studies [9-14]; these studies resulted in documenting functional [9] and anatomical (partial and complete) [2,10-14] closure of VSDs (Figure 1 and Figure 2). These result suggested that spontaneous closure of physiologically advantageous VSDs occurs as often as in isolated VSDs. The mechanisms of closure is also similar to that seen in isolated VSDs. VSD closures were documented to take place in utero, during infancy, childhood, and adolescence and continue through adulthood. It was concluded that there is a great natural tendency for spontaneous closure of VSDs whether it be an isolated defect or is a component of a complex heart defect [10-16].
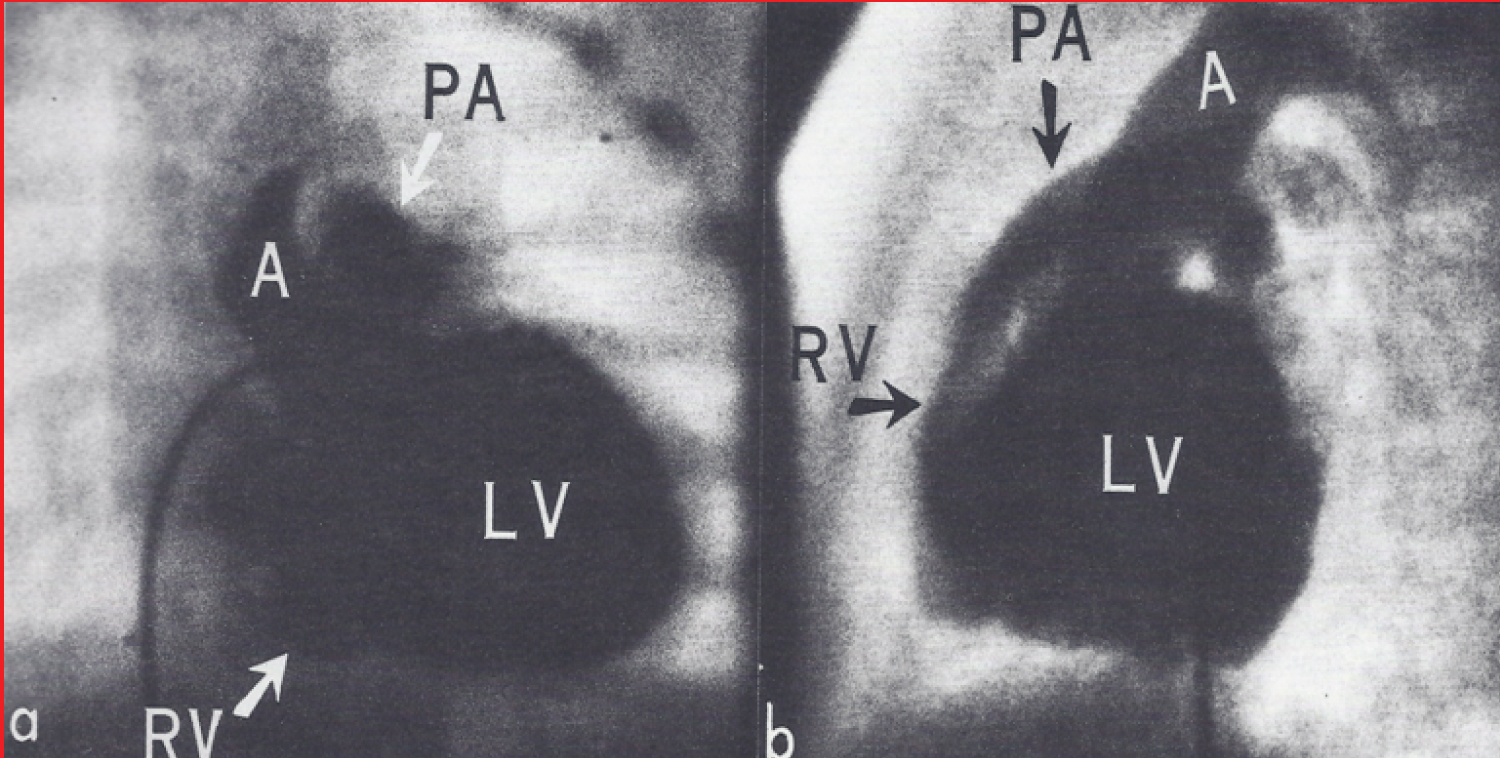 Figure 1: a) Selective left ventricular (LV) cineangiogram in antero-posterior and b) Lateral views at the age 5.5 months, demonstrating opacification of the right ventricle (RV) via a ventricular septal defect (not labeled) [2].
Figure 1: a) Selective left ventricular (LV) cineangiogram in antero-posterior and b) Lateral views at the age 5.5 months, demonstrating opacification of the right ventricle (RV) via a ventricular septal defect (not labeled) [2].
A: Aorta; PA: Pulmonary Artery.
View Figure 1
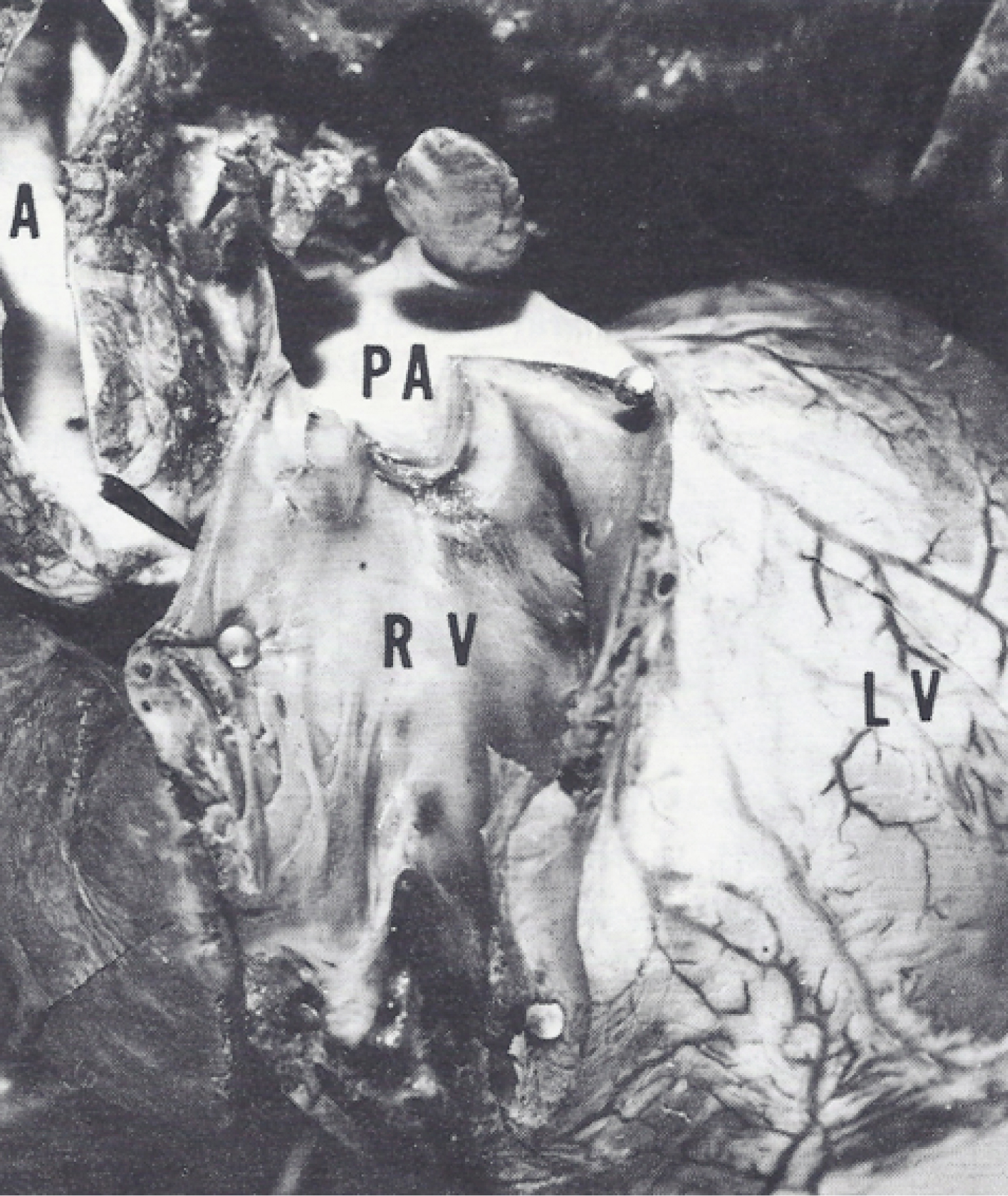 Figure 2: Photograph of the heart with the right ventricle (RV) open at autopsy at the age of 2.5 years, demonstrating no evidence of the previously seen (Figure 1) ventricular septal defect [2].
Figure 2: Photograph of the heart with the right ventricle (RV) open at autopsy at the age of 2.5 years, demonstrating no evidence of the previously seen (Figure 1) ventricular septal defect [2].
A: Aorta; PA: Main pulmonary artery.
View Figure 2
Spontaneous closure of VSD in children with tricuspid atresia and normally related great arteries (Type I) produces decreased pulmonary blood flow and requires surgical intervention earlier than otherwise needed. We recommended that a Blalock-Taussig (BT) shunt (preferably on the left side) is carried out instead of a Glenn anastomosis (the concept of bidirectional Glenn has not yet been crystallized as of that time) because VSD closure following Glenn will result in no blood flow into the left pulmonary artery (Figure 3). If further palliation is required prior to Fontan procedure, a BT shunt on the right side, Glenn anastomosis, or enlargement of the VSD may be entertained. In subjects with tricuspid atresia and transposition of the great arteries (Type II), VSD closure produces left ventricular outflow obstruction. Therefore, it is highly important that the size of the VSD is appraised before planning for Fontan. If the VSD is small, enlarging the VSD surgically or bypassing the VSD and the right ventricle via a pulmonary artery-to-ascending aorta anastomosis (Damus-Kaye-Stansel) or by a left ventricle-descending aorta conduit (Figure 4) should be considered [10-14]. In addition to tricuspid atresia and double outlet right ventricle, spontaneous closure of VSDs was observed in patients with tetralogy of Fallot, pulmonary atresia with VSD and transposition of the great arteries with intact atrial septum, and double inlet left ventricle with ventriculo-arterial discordance [15-17].
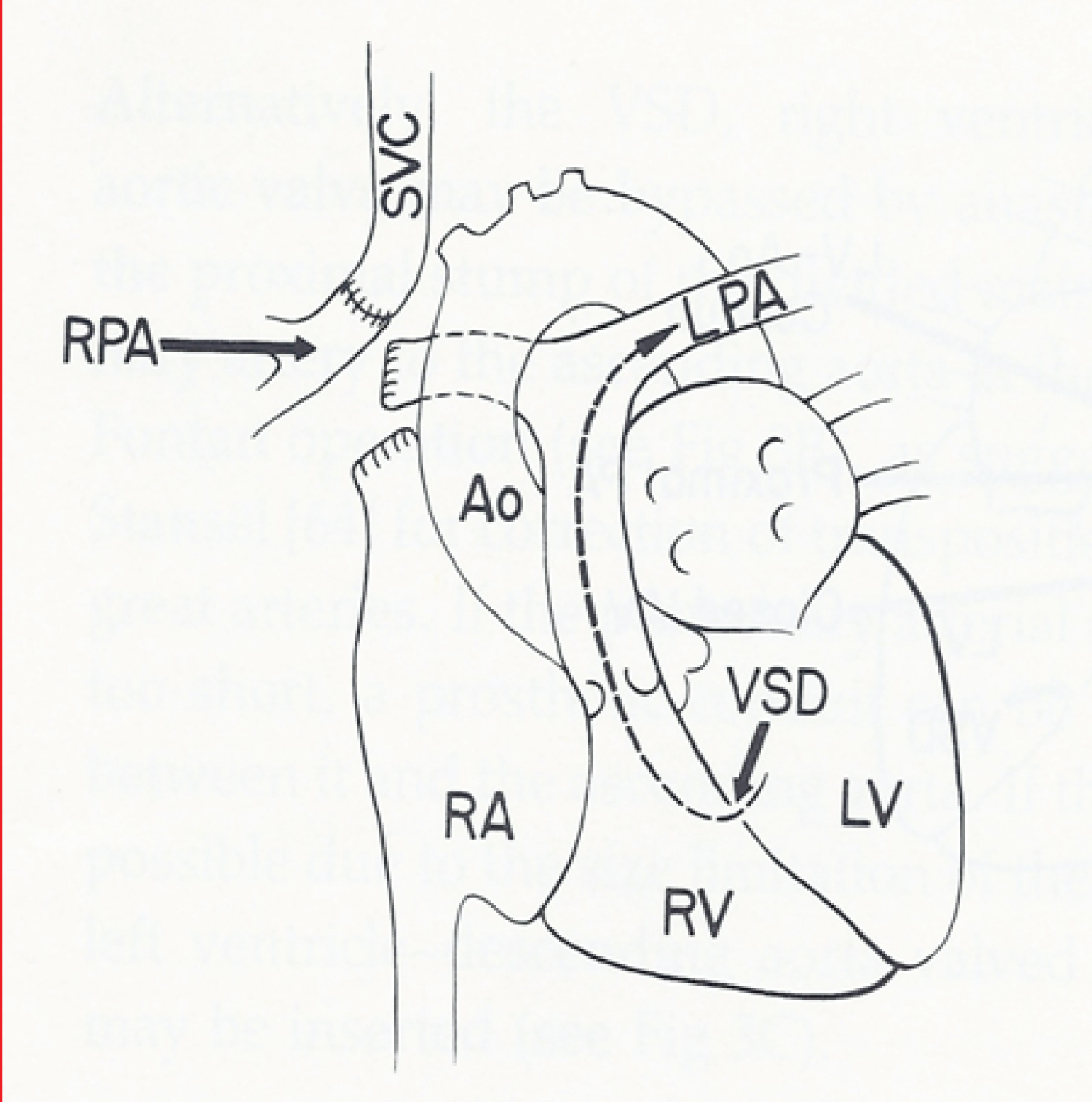 Figure 3: Line drawing of classic Glenn anastomosis connecting the superior vena cava (SVC) with the right pulmonary artery (RPA). Should the ventricular septal defect (VSD) close spontaneously, the left pulmonary artery (LPA) will be without blood flow [13].
Figure 3: Line drawing of classic Glenn anastomosis connecting the superior vena cava (SVC) with the right pulmonary artery (RPA). Should the ventricular septal defect (VSD) close spontaneously, the left pulmonary artery (LPA) will be without blood flow [13].
Ao: Aorta; LV: Left Ventricle; RA: Right Atrium; RV: Right Ventricle.
View Figure 3
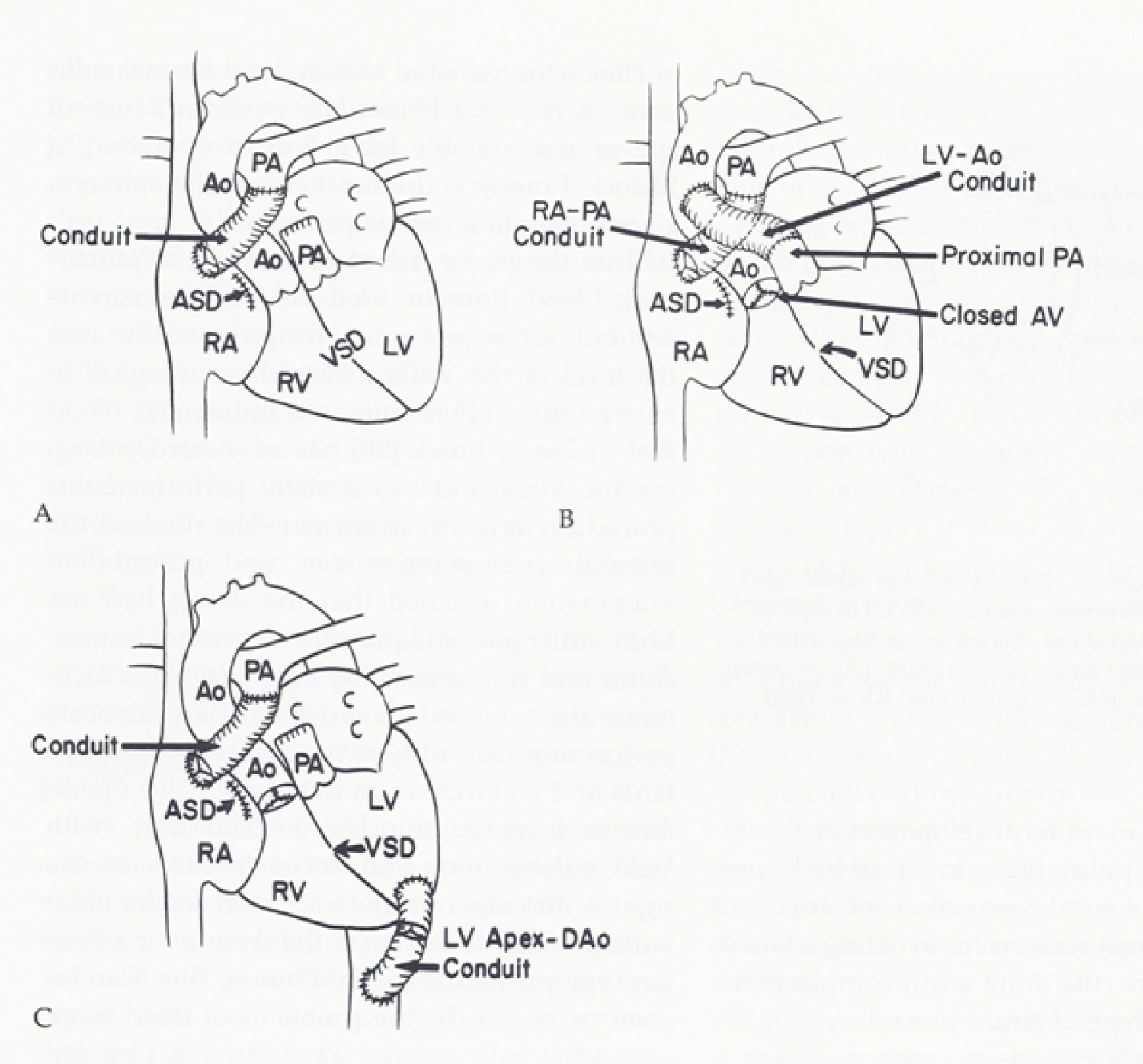 Figure 4: A) Line drawing illustrating atrio-pulmonary type of Fontan for patients with tricuspid atresia with transposition of the great arteries. The left ventricular (LV) blood flows via the ventricular septal defect (VSD) and right ventricle (RV) into the aorta (Ao); B) If the VSD is small and restrictive, causing "subaortic" obstruction, this obstruction may be bypassed by connecting the proximal stump of the divided pulmonary artery (PA) to the Ao directly or via a non-valved conduit; C) An alternate approach is to connect the LV apex to the descending aorta (DAo) [10,13].
Figure 4: A) Line drawing illustrating atrio-pulmonary type of Fontan for patients with tricuspid atresia with transposition of the great arteries. The left ventricular (LV) blood flows via the ventricular septal defect (VSD) and right ventricle (RV) into the aorta (Ao); B) If the VSD is small and restrictive, causing "subaortic" obstruction, this obstruction may be bypassed by connecting the proximal stump of the divided pulmonary artery (PA) to the Ao directly or via a non-valved conduit; C) An alternate approach is to connect the LV apex to the descending aorta (DAo) [10,13].
ASD: Atrial Septal Defect; RA: Right Atrium.
View Figure 4
The interest of the author in the phenomenon of spontaneous closure of physiologically advantageous VSDs resulted in examining this issue in patients with tricuspid atresia [9-14]. While studying these issues, the author's interest in other features of tricuspid atresia emerged. The ensuing investigations resulted in the publication of a number of papers and book chapters [18-32] including two books [33,34]. In this section of the book, terminology [35-37]; unified classification (Figure 5 and Figure 6) [38-40]; description of rare types of tricuspid atresia (Figure 7, Figure 8, Figure 9, Figure 10, Figure 11 and Figure 12) [41-44]; were reviewed.
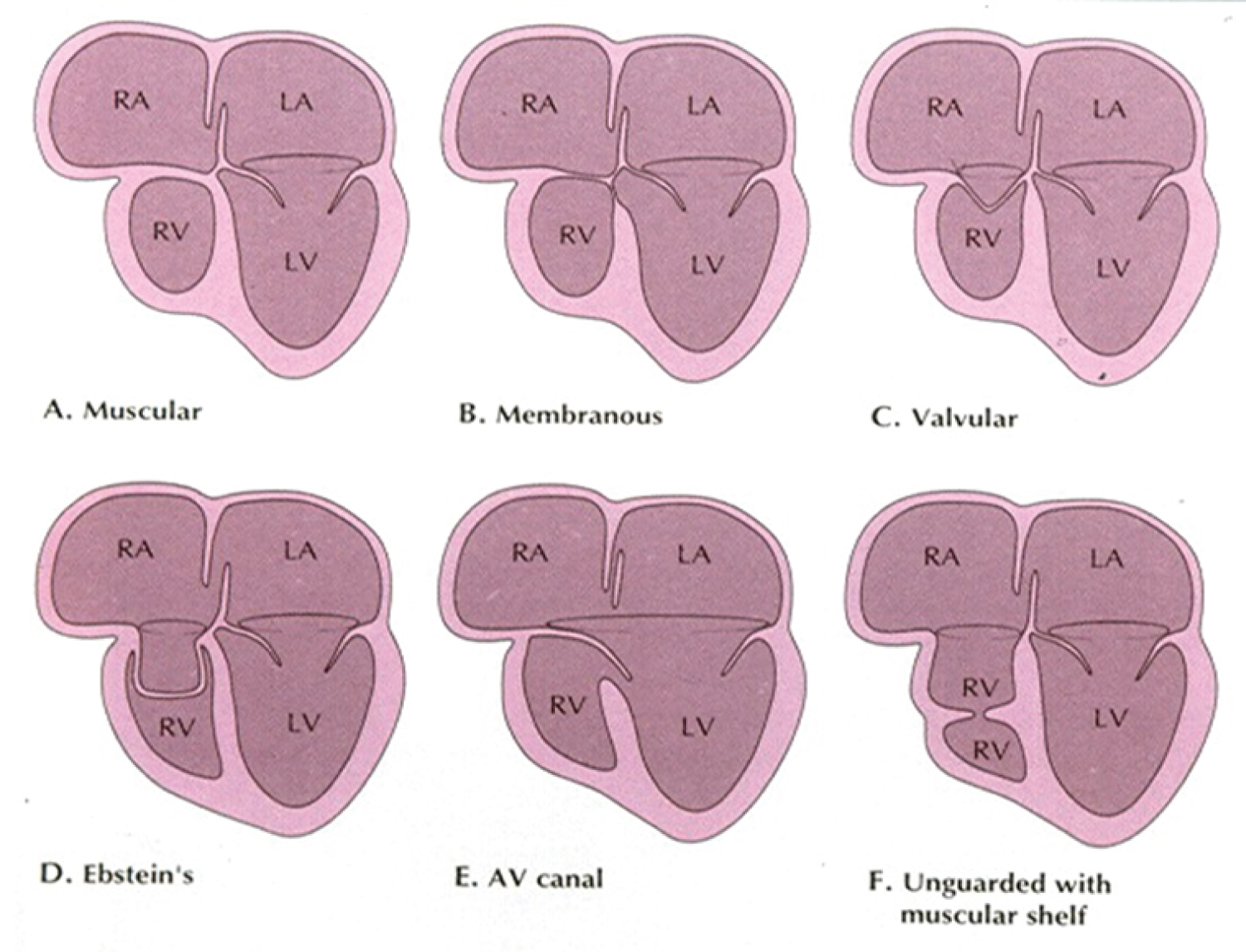 Figure 5: A) Classification based on morphology of the tricuspid valve is shown diagrammatically. The classic muscular type is shown with a thick band of tissue where the tricuspid valve should be; B) Membranous type; C) Valvular type with fused valve leaflets; D) Ebstein's type with fused valve cusps along with Ebstein type of downward displacement of the tricuspid valve; E) Atrioventricular (AV) canal (septal) type with common AV leaflet sealing off the right ventricle (RV) from the right atrium (RA) and F) Unguarded muscular shelf type where the RV is divided into inlet and outlet portions by a muscular shelf [19].
Figure 5: A) Classification based on morphology of the tricuspid valve is shown diagrammatically. The classic muscular type is shown with a thick band of tissue where the tricuspid valve should be; B) Membranous type; C) Valvular type with fused valve leaflets; D) Ebstein's type with fused valve cusps along with Ebstein type of downward displacement of the tricuspid valve; E) Atrioventricular (AV) canal (septal) type with common AV leaflet sealing off the right ventricle (RV) from the right atrium (RA) and F) Unguarded muscular shelf type where the RV is divided into inlet and outlet portions by a muscular shelf [19].
LA: Left Atrium; LV: Left Ventricle
View Figure 5
 Figure 6: Unified classification based on associated cardiac defects [19,38].
View Figure 6
Figure 6: Unified classification based on associated cardiac defects [19,38].
View Figure 6
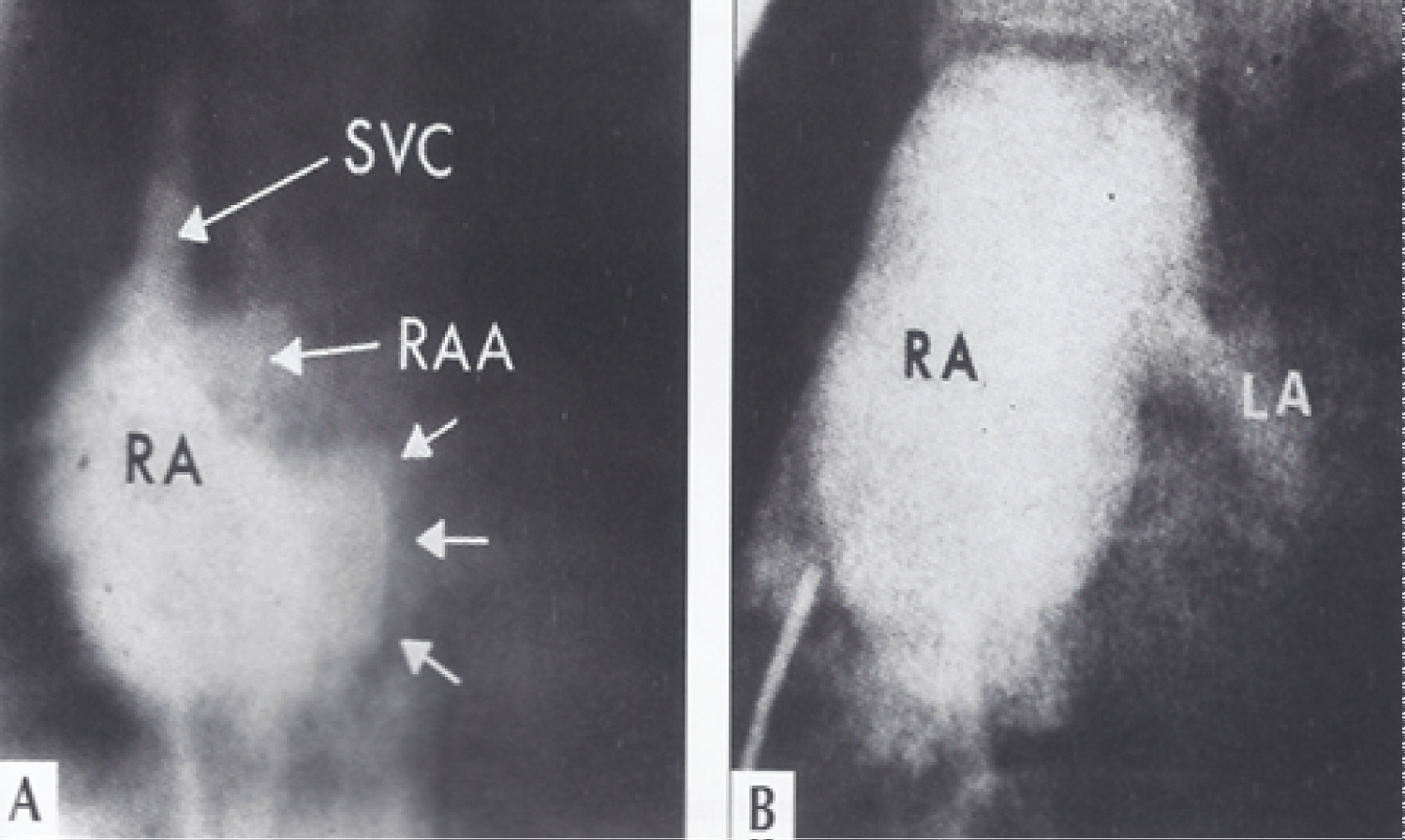 Figure 7: A) Selected right atrial (RA) cineangiographic frame in postero-anterior view prior to opacification of the left atrium (LA) in a patient with tricuspid atresia with Ebstein's malformation. The contrast material delineates the atretic tricuspid valve and is shown by small unlabelled arrows. This is rounded and extends more to the left than in other tricuspid atresia cases; B) Right atrial angiographic frame from a patient with classic tricuspid atresia is shown for comparison with figure A. This frame is frozen prior to full opacification of the LA [41].
Figure 7: A) Selected right atrial (RA) cineangiographic frame in postero-anterior view prior to opacification of the left atrium (LA) in a patient with tricuspid atresia with Ebstein's malformation. The contrast material delineates the atretic tricuspid valve and is shown by small unlabelled arrows. This is rounded and extends more to the left than in other tricuspid atresia cases; B) Right atrial angiographic frame from a patient with classic tricuspid atresia is shown for comparison with figure A. This frame is frozen prior to full opacification of the LA [41].
RAA: Right Atrial Appendage; SVC: Superior Vena Cava.
View Figure 7
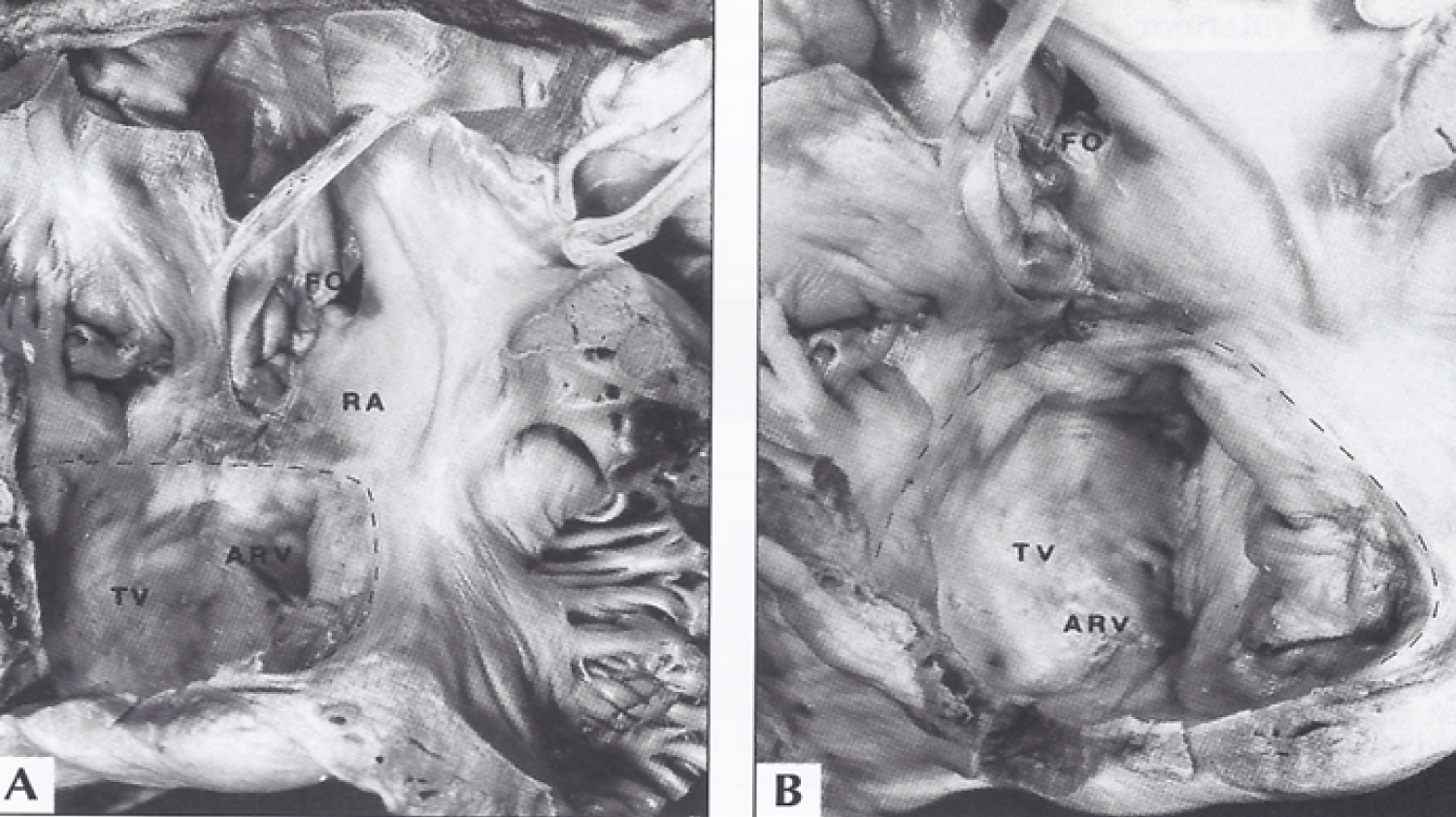 Figure 8: A) The interior of the right atrium and of the atrialized right ventricle (ARV) in a patient with tricuspid atresia with Ebstein's malformation are shown. B) This is a close-up of A. The interrupted line demarcates the true tricuspid valve annulus. The tricuspid valve (TV) leaflets are displaced downward (Ebstein's malformation) and fused, incorporating a part of the right ventricle into the right atrium (RA) [41].
Figure 8: A) The interior of the right atrium and of the atrialized right ventricle (ARV) in a patient with tricuspid atresia with Ebstein's malformation are shown. B) This is a close-up of A. The interrupted line demarcates the true tricuspid valve annulus. The tricuspid valve (TV) leaflets are displaced downward (Ebstein's malformation) and fused, incorporating a part of the right ventricle into the right atrium (RA) [41].
FO: Fossa Ovalis.
View Figure 8
 Figure 9: Selected two-dimensional, subcostal, four-chamber echocardiographic frames with an open (a) and closed (b) atrioventricular valve. Line drawings on the right of a and b are made for greater clarity and for labeling. A large ostium primum atrial septal defect (10 ASD) is shown in a. When the large atrioventricular valve leaflet is open (a), it completely closes the right ventricle (RV) from the right atrium (RA) and the ventricular septal defect (VSD) and allows the emptying of blood from both atria into the left ventricle (LV). When the atrioventricular valve leaflet is closed (b), it continues to occlude the RV from the RA while allowing the VSD to freely communicate between the RV and LV [42].
Figure 9: Selected two-dimensional, subcostal, four-chamber echocardiographic frames with an open (a) and closed (b) atrioventricular valve. Line drawings on the right of a and b are made for greater clarity and for labeling. A large ostium primum atrial septal defect (10 ASD) is shown in a. When the large atrioventricular valve leaflet is open (a), it completely closes the right ventricle (RV) from the right atrium (RA) and the ventricular septal defect (VSD) and allows the emptying of blood from both atria into the left ventricle (LV). When the atrioventricular valve leaflet is closed (b), it continues to occlude the RV from the RA while allowing the VSD to freely communicate between the RV and LV [42].
Ap: Apex; AtTV: Atretic Tricuspid Valve; Ba: Base; L: Left; LA: Left Atrium; R: Right
View Figure 9
 Figure 10: Selected right atrial (RA) angiographic frame in postero-anterior view, demonstrating that the floor of the right atrium is formed by one of the leaflets of the atrioventricular valve; this is marked by a large arrow. The contrast material exited the RA via an ostium primum atrial septal defect, shown by small arrows, with subsequent opacification of the left ventricle (LV) [42].
LA: Left Atrium.
View Figure 10
Figure 10: Selected right atrial (RA) angiographic frame in postero-anterior view, demonstrating that the floor of the right atrium is formed by one of the leaflets of the atrioventricular valve; this is marked by a large arrow. The contrast material exited the RA via an ostium primum atrial septal defect, shown by small arrows, with subsequent opacification of the left ventricle (LV) [42].
LA: Left Atrium.
View Figure 10
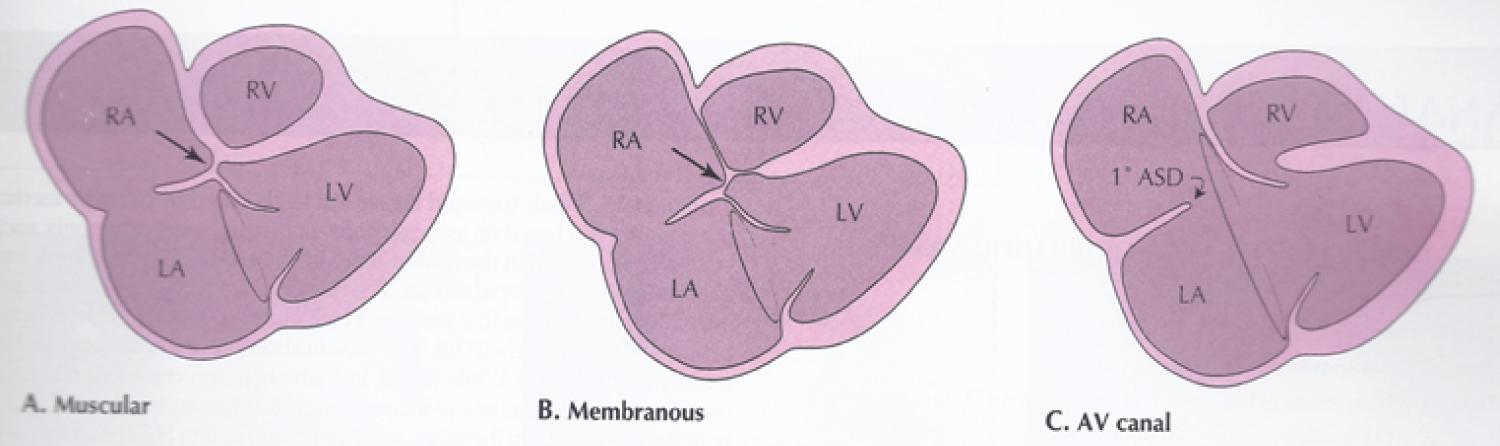 Figure 11: Line drawings demonstrating two-dimensional echocardiographic appearances in a subcostal four-chamber view of the muscular (A), membranous (B) and atrioventricular canal (C) variants of tricuspid atresia. A) The atretic tricuspid valve is represented by a thick band of echoes between the right atrium (RA) and the small right ventricle (RV) in the muscular type; B) The tricuspid valve is represented by a thin line in the membranous type. Note that crux of the heart (arrows in A and B) is clearly seen in both these types (A and B). The attachment of the anterior leaflet of the detectable atrioventricular valve to the left side of the interatrial septum is evident; C) In the atrioventricular canal type of tricuspid atresia, the anterior leaflet of the detectable atrioventricular canal is attached to the anterior wall of the heart, occluding the right ventricle from the right atrium, and allows the exit of blood from both atria into the left ventricle (LV). The crux cordis and the atrioventricular portion of the inter-ventricular septum are not seen [19,42].
View Figure 11
Figure 11: Line drawings demonstrating two-dimensional echocardiographic appearances in a subcostal four-chamber view of the muscular (A), membranous (B) and atrioventricular canal (C) variants of tricuspid atresia. A) The atretic tricuspid valve is represented by a thick band of echoes between the right atrium (RA) and the small right ventricle (RV) in the muscular type; B) The tricuspid valve is represented by a thin line in the membranous type. Note that crux of the heart (arrows in A and B) is clearly seen in both these types (A and B). The attachment of the anterior leaflet of the detectable atrioventricular valve to the left side of the interatrial septum is evident; C) In the atrioventricular canal type of tricuspid atresia, the anterior leaflet of the detectable atrioventricular canal is attached to the anterior wall of the heart, occluding the right ventricle from the right atrium, and allows the exit of blood from both atria into the left ventricle (LV). The crux cordis and the atrioventricular portion of the inter-ventricular septum are not seen [19,42].
View Figure 11
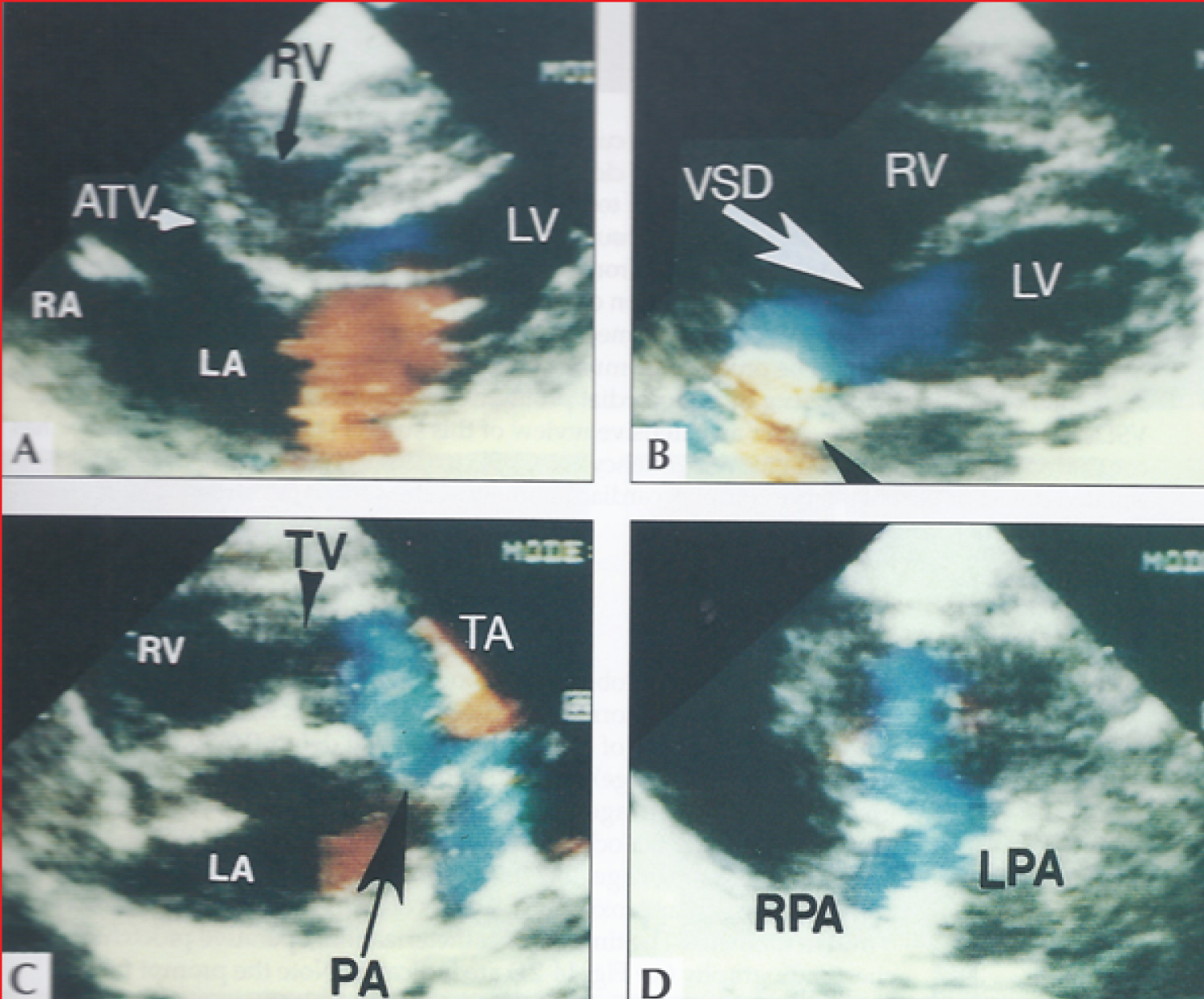 Figure 12: Selected video frames from a two-dimensional echocardiographic and color Doppler study demonstrating a) An atretic tricuspid valve (ATV) between the right atrium (RA) and right ventricle (RV) and blood flow from the left atrium (LA) into the left ventricle (LV) across the mitral valve. The RV (arrow) is very small and hypoplastic; b) LV and RV with a large ventricular septal defect (VSD) below the truncus arteriosus (TA). Turbulent flow across the truncal valve suggests truncal valve stenosis; c) The origin of the pulmonary artery (PA) from the TA by color flow (arrow), and d) The division of right (RPA) and left (LPA) pulmonary arteries from the PA (not labeled in d) in a short-axis view [43].
Figure 12: Selected video frames from a two-dimensional echocardiographic and color Doppler study demonstrating a) An atretic tricuspid valve (ATV) between the right atrium (RA) and right ventricle (RV) and blood flow from the left atrium (LA) into the left ventricle (LV) across the mitral valve. The RV (arrow) is very small and hypoplastic; b) LV and RV with a large ventricular septal defect (VSD) below the truncus arteriosus (TA). Turbulent flow across the truncal valve suggests truncal valve stenosis; c) The origin of the pulmonary artery (PA) from the TA by color flow (arrow), and d) The division of right (RPA) and left (LPA) pulmonary arteries from the PA (not labeled in d) in a short-axis view [43].
TV: Truncal Valve Leaflets.
View Figure 12
Also discussed were: enumeration of demographic [45,46], electrocardiographic [47-49], and echocardiographic features [50-52]; description of new findings of left to right atrial shunt in tricuspid atresia (Figure 13 and Figure 14) [53]; cardiac catheterization [54,55]; principles of palliation[56,57]; role of percutaneous interventions [58,59]; and insights into surgical approaches [60-62].
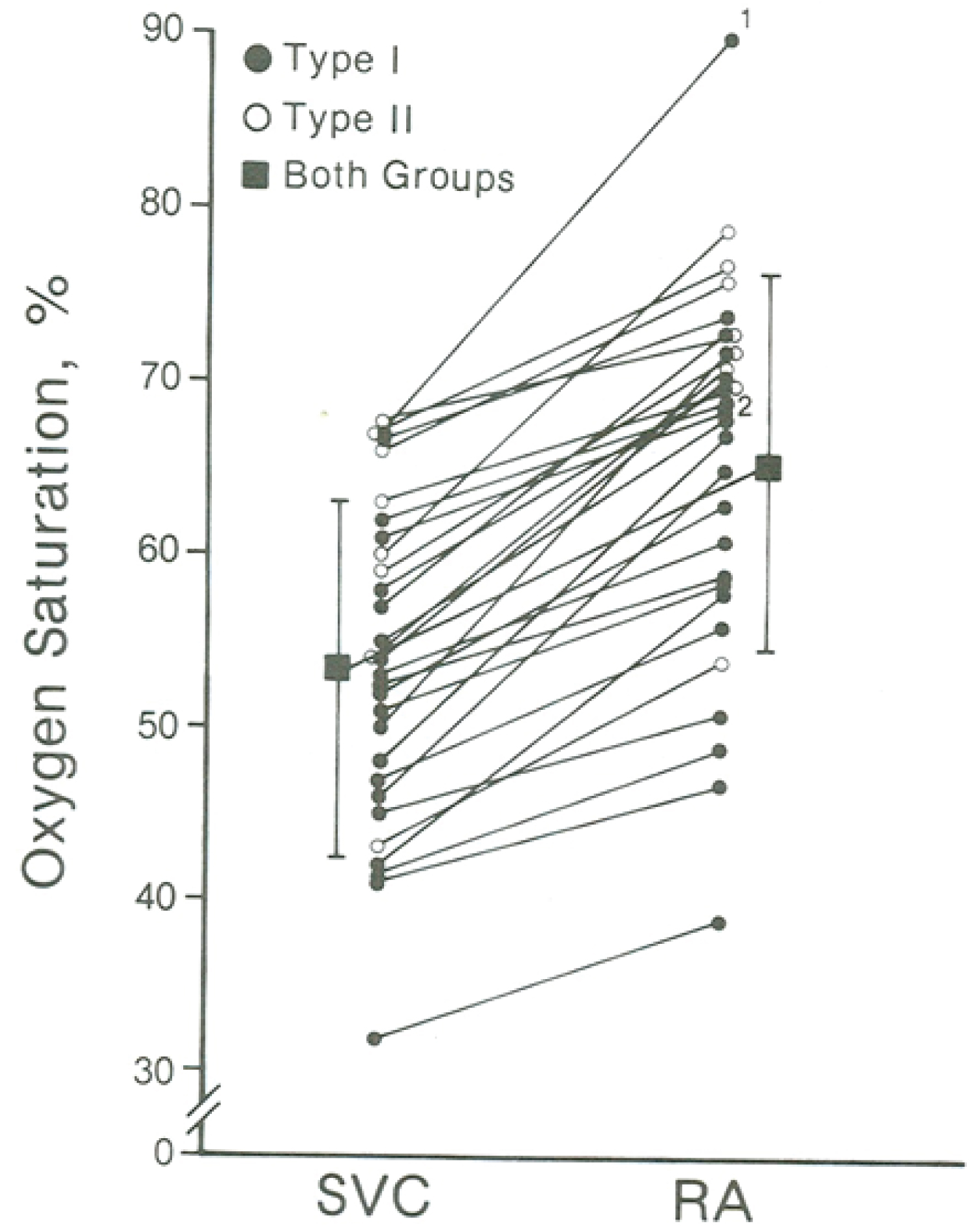 Figure 13: Oxygen saturation data of tricuspid atresia patients with left-to-right shunt. The superior vena caval (SVC) and the right atrial (RA) saturations in % are shown. Each saturation is marked as closed circles in Type I (normally related great arteries) patients and as open circles in Type II (transposition of the great arteries) patients. There is a statistically significant (p < 001) increase in oxygen saturation from the SVC to RA (square). The magnitude of the increase in shunt between Type I and Type II patients is not significant (p > 0.1) (not shown) [53].
View Figure 13
Figure 13: Oxygen saturation data of tricuspid atresia patients with left-to-right shunt. The superior vena caval (SVC) and the right atrial (RA) saturations in % are shown. Each saturation is marked as closed circles in Type I (normally related great arteries) patients and as open circles in Type II (transposition of the great arteries) patients. There is a statistically significant (p < 001) increase in oxygen saturation from the SVC to RA (square). The magnitude of the increase in shunt between Type I and Type II patients is not significant (p > 0.1) (not shown) [53].
View Figure 13
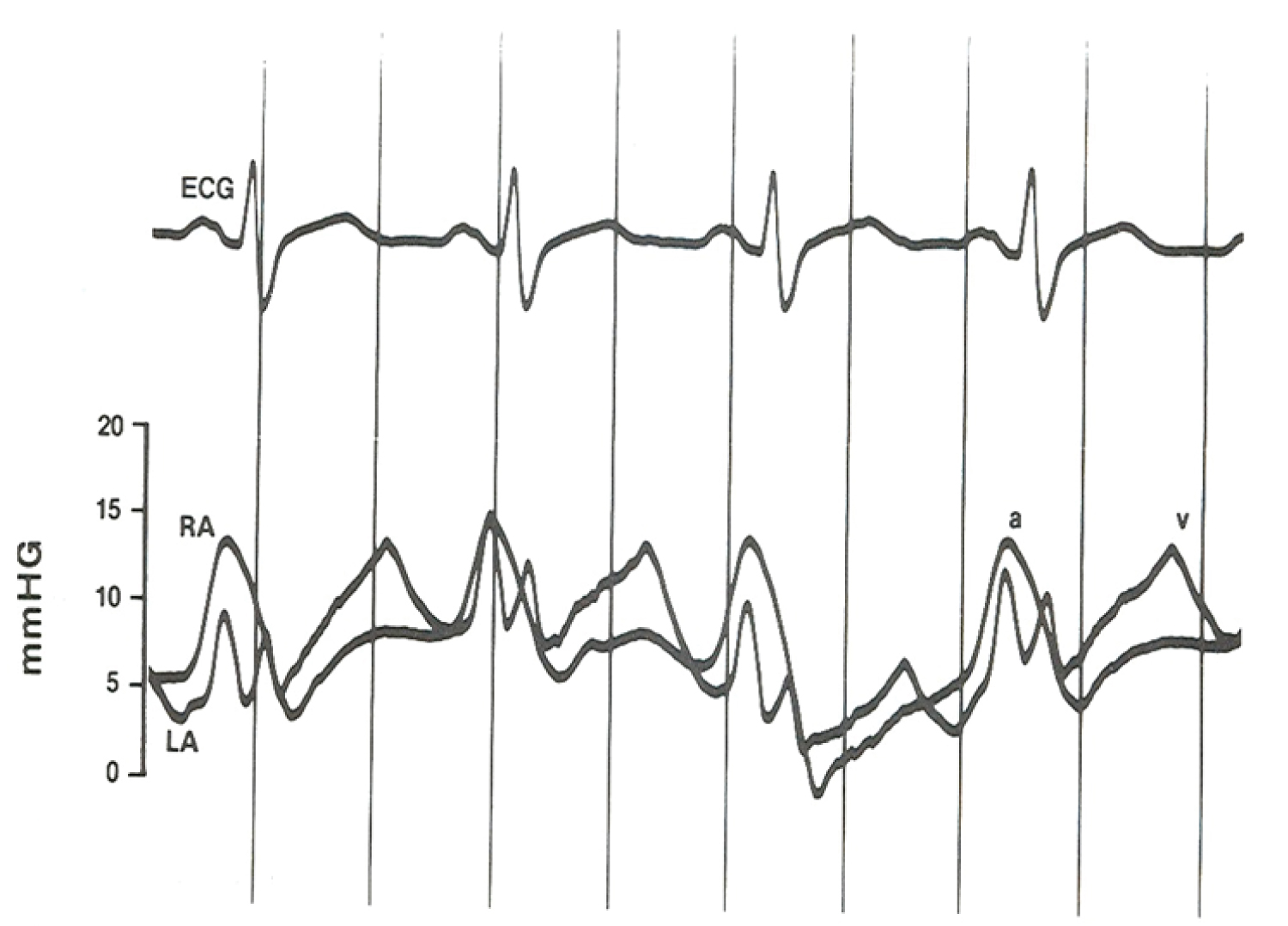 Figure 14: Pressure tracings from the left atrium (LA) and right atrium (RA) recorded simultaneously via high fidelity miniature transducers mounted within the catheter 5 cm apart, one placed in the LA and the other in the RA. These tracings demonstrate higher pressure in the RA than the LA during atrial systole (a wave) and lower pressures in the RA than LA during atrial diastole (v wave); the later may have been responsible for the left-to-right shunt. It may be concluded that these instantaneous pressure differences between atria are likely to explain the left-to-right shunt in tricuspid atresia [53].
ECG: Electrocardiogram
View Figure 14
Figure 14: Pressure tracings from the left atrium (LA) and right atrium (RA) recorded simultaneously via high fidelity miniature transducers mounted within the catheter 5 cm apart, one placed in the LA and the other in the RA. These tracings demonstrate higher pressure in the RA than the LA during atrial systole (a wave) and lower pressures in the RA than LA during atrial diastole (v wave); the later may have been responsible for the left-to-right shunt. It may be concluded that these instantaneous pressure differences between atria are likely to explain the left-to-right shunt in tricuspid atresia [53].
ECG: Electrocardiogram
View Figure 14
Management of cyanotic congenital heart defects (CHDs) was detailed. Whereas the need for addressing the acyanotic CHD patients is to a great degree dependant on the severity of the lesion, the majority of cyanotic CHDs need intervention, typically by a variety of surgical procedures. At first, the pathophysiological consequences of cyanotic CHD were reviewed [63]. Then, the discussion of general management cyanotic CHDs [64,65]. This is followed by a brief description of each of these cyanotic CHDs and then their management.
Different types of tetralogy of Fallot (TOF) babies necessitate different types of surgical procedures [65-68]. Most of conventional TOF infants are either acyanotic or minimally cyanotic at birth and may not need surgical intervention in the neonatal period. Hyper cyanotic spells may occur in TOF patients, mostly during infancy. The possible etiology and management of these spells were described [63,66,68]. Total surgical correction involves closure of VSD allowing the left ventricle (LV) pumping into the aorta, and alleviating the infundibular, valvar and pulmonary artery obstruction with or without trans-annular patch, all under cardiopulmonary bypass; this is generally undertaken between three to 12 months of age. Pulmonary valve-sparing surgery is more frequently performed at the present time. Some of these babies may need initial palliation, mostly by a modified BT shunt. Some babies may be benefitted by balloon pulmonary valvuloplasty in place of modified BT shunts to augment the pulmonary blood flow.
The early management of TOF with pulmonary atresia who are ductal dependent is by prompt intravenous administration of PGE1 to keep the ductus open. After the infant is stabilized, a stable way to provide pulmonary blood flow should be instituted. In infants whose defects cannot be completely corrected in the newborn period, a modified BT shunt using an interposition Gore-Tex graft between right or left subclavian artery and the ipsilateral pulmonary artery is current surgical procedure of choice. Ductal stents are used at some institutions instead. Total surgical correction with closure of VSD and pulmonary valvotomy with or without trans-annular patch or insertion of a right ventricle to pulmonary artery valved conduit (usually a homograft) under cardio-pulmonary bypass is performed when the baby is between six and twelve months of age [65,67,69-71].
The early management of TOF with pulmonary atresia who are ductal dependent is by prompt intravenous administration of PGE1 to keep the ductus open. After the infant is stabilized, a stable way to provide pulmonary blood flow should be instituted. In infants whose defects cannot be completely corrected in the newborn period, a modified BT shunt using an interposition Gore-Tex graft between right or left subclavian artery and the ipsilateral pulmonary artery is current surgical procedure of choice. Ductal stents are used at some institutions instead. Total surgical correction with closure of VSD and pulmonary valvotomy with or without trans-annular patch or insertion of a right ventricle to pulmonary artery valved conduit (usually a homograft) under cardio-pulmonary bypass is performed when the baby is between six and twelve months of age [65,67,69-71].
Symptomatic newborn babies with TOF and syndrome of absent pulmonary valve need ventilatory support to stabilize the patient; this is followed by total surgical correction using cardiopulmonary bypass. Closure of the VSD, relief of PS by a trans-annular pericardial patch as needed, and partial resection and plastic repair of aneurysmally dilated pulmonary arteries are undertaken [65,67,74].
Infants with transposition of the great arteries with intact ventricular septum as well as the babies with VSD require arterial switch (Jatene) operation whereas babies with both VSD and pulmonary stenosis (PS) are managed with Rastelli procedure [65-68,75]. These surgical procedures may have to be preceded by prostaglandin infusion and/or balloon atrial septostomy in some infants [65-68,75]. Babies with tricuspid atresia need palliative intervention either with a modified BT shunt or banding of the pulmonary artery, as the case may be with subsequent staged Fontan correction consisting of bidirectional Glenn and fenestrated Fontan with extra-cardiac conduit [26,28,62,65]. Infants with total anomalous pulmonary venous connection are treated by anastomosing the common pulmonary venous confluence with the left atrium either electively in non-obstructed types or as an emergent surgery in the obstructed varities [65,73,76,77]. Infants with truncus arteriosus are managed wit surgical intervention which includes closure of VSD coupled with right ventricle to pulmonary artery conduit [65,76,78-80].
The medical and surgical treatment of other CHDs, namely, hypoplastic left heart syndrome [72,73,81], pulmonary atresia with intact ventricular septum [65,69,72,73,81,82], double-outlet right ventricle [65,76,83], univentricular hearts including double-inlet left ventricle [17,34,65,72,76], corrected transposition of the great arteries [65,84-86], Ebstein's anomaly of the tricuspid valve [65,85,87-89], pulmonary atresia without intact ventricular septum (with VSD) [17,34,65,72,76,84-86], mitral atresia with normal aortic root [34,65,90,91], interrupted aortic arch [65,92], and dextrocardia and heterotaxy syndromes (asplenia and polysplenia) [65,93-96] was also discussed. The presently used medical, trans-catheter and surgical procedures to treat cyanotic CHD are safe and effective and can be performed at a fairly low risk.
The contributions of the author with regard to adult CHD issues, namely infective endocarditis, trans-catheter closure of PDA, trans-catheter occlusion of ASDs, trans-catheter closure of ASD/PFO for prevention of recurrence of paradoxical embolism, closure of ASDs/PFOs with right-to-left shunt in children who had prior cardiac surgery, percutaneous closure of aorto-pulmonary window, trans-catheter management of platypnea-orthodeoxia syndrome, and balloon dilatation of CoA, PS, and aortic stenosis in adults were discussed and referenced in Parts I and II of the book summary and will not be repeated. Then, a discussion of "What an adult cardiologist should know about cyanotic CHD" was presented [97,98]. The prevalence of CHDs in adult subjects has increased during the last two decades such that there are more adults with CHD than children at this time. Advances in the diagnosis and treatment of CHD during infancy and childhood seem to be the reason for this phenomenon. Most of these patients are likely to be those who previously had corrective or palliative surgery during infancy and childhood, though rarely uncorrected CHDs may present in adulthood. Pathophysiologic effects of right to left shunt associated with cyanotic CHD were discussed. Brief narrative of the anatomy of the most common cyanotic CHDs such as tetralogy of Fallot, transposition of the great arteries, truncus arteriosus, total anomalous pulmonary venous connection and tricuspid atresia was reviewed and their management. The nature of the residual defects, their long-term effects and management strategies for each of the above CHDs during adulthood were addressed. Adults who did not have surgical correction in childhood should have surgical correction; however, there is a higher risk for surgery than that seen in children [97,98].
This section addresses miscellaneous topics that could not be included in the earlier sections of the book. These topics, namely, cor pulmonale as a complication following cerebral ventriculo-atrial shunts [99], physiological basis of diuretic medications [100], CHD in babies with the de Lange syndrome [101], preventive aspects of all types of heart disease in infants and children [102-106], obstruction of the superior vena cava in total anomalous pulmonary venous connection [107], early identification of the newborn with serious heart disease [108-111], infective endocarditis [112-114], and utility of continuous positive airway pressure in the differential diagnosis of cardiac from pulmonary causes of cyanosis in the neonate (Figure 15 ) [115] were discussed.
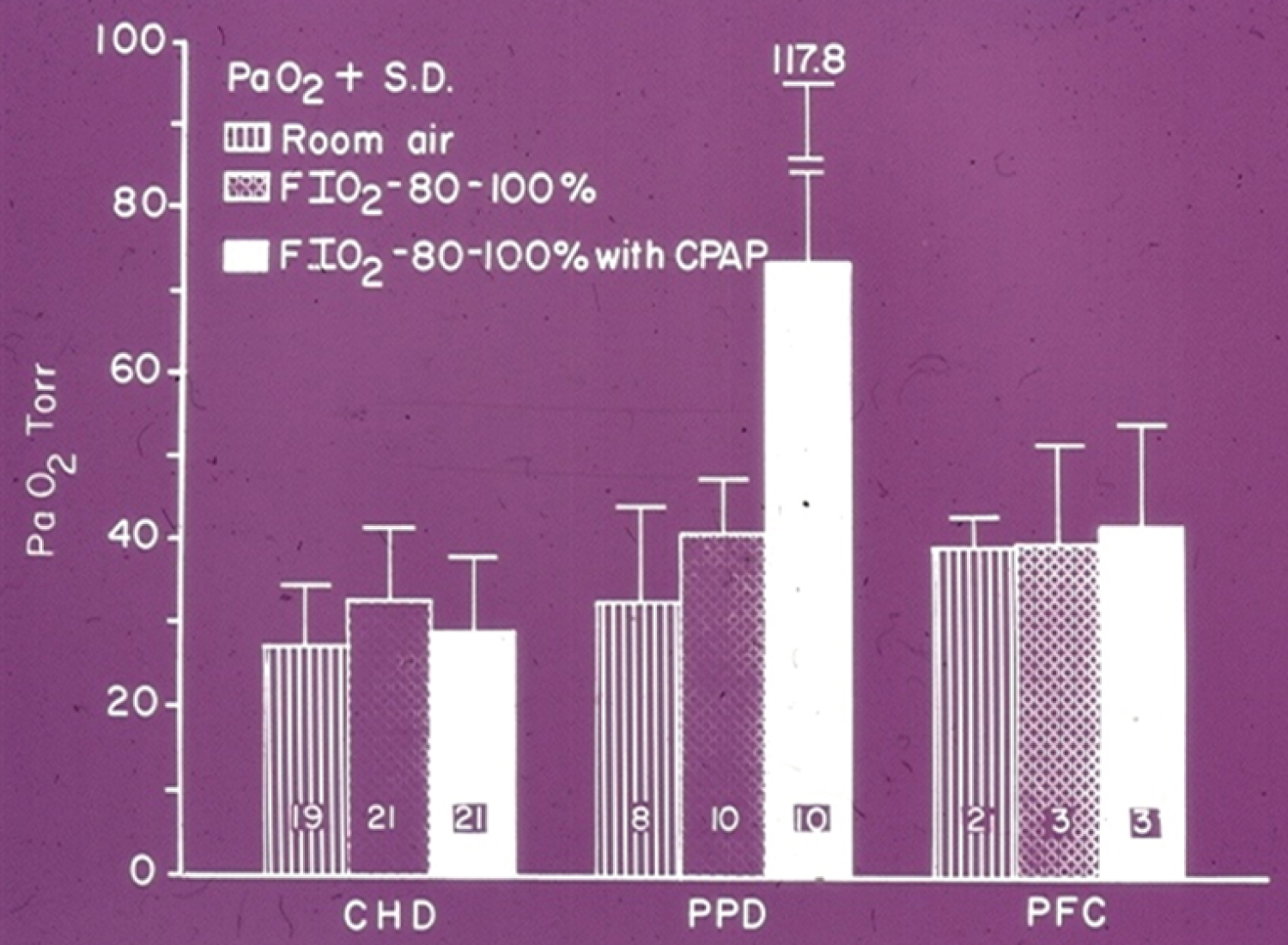 Figure 15: The PaO2 is expressed as the mean + standard deviation. The number of infants in each group is shown in each bar. There was no change in PaO2 from F1O2 of 0.21-0.4 to F1O2 of 0.8-1.0 in all three groups. With additional CPAP the PaO2 did not change in babies with congenital heart defects (CHD) and persistent fetal circulation (PFC). However, a significant increase in PaO2 occurred in the pulmonary parenchymal disease (PD) group after CPAP [115].
View Figure 15
Figure 15: The PaO2 is expressed as the mean + standard deviation. The number of infants in each group is shown in each bar. There was no change in PaO2 from F1O2 of 0.21-0.4 to F1O2 of 0.8-1.0 in all three groups. With additional CPAP the PaO2 did not change in babies with congenital heart defects (CHD) and persistent fetal circulation (PFC). However, a significant increase in PaO2 occurred in the pulmonary parenchymal disease (PD) group after CPAP [115].
View Figure 15
Other topics reviewed were: polysplenia syndrome [93], present status of surgery in CHD [116,117], systematic approach to differential diagnosis of dextrocardia [93-96,118], vasodilator therapy for heart failure in pediatric practice [119-122], a new complication of chylopericardium in babies who had BT anastomosis [123-125], scimitar syndrome [126], management of syndrome of absent pulmonary valve [74,127,128], mitral valve prolapse [129,130], anticoagulant therapy in children with prosthetic valves (Figure 16) [131], long-term oral diazoxide therapy for pulmonary vascular obstructive disease associated with CHDs (Figure 17 and Figure 18) [132].
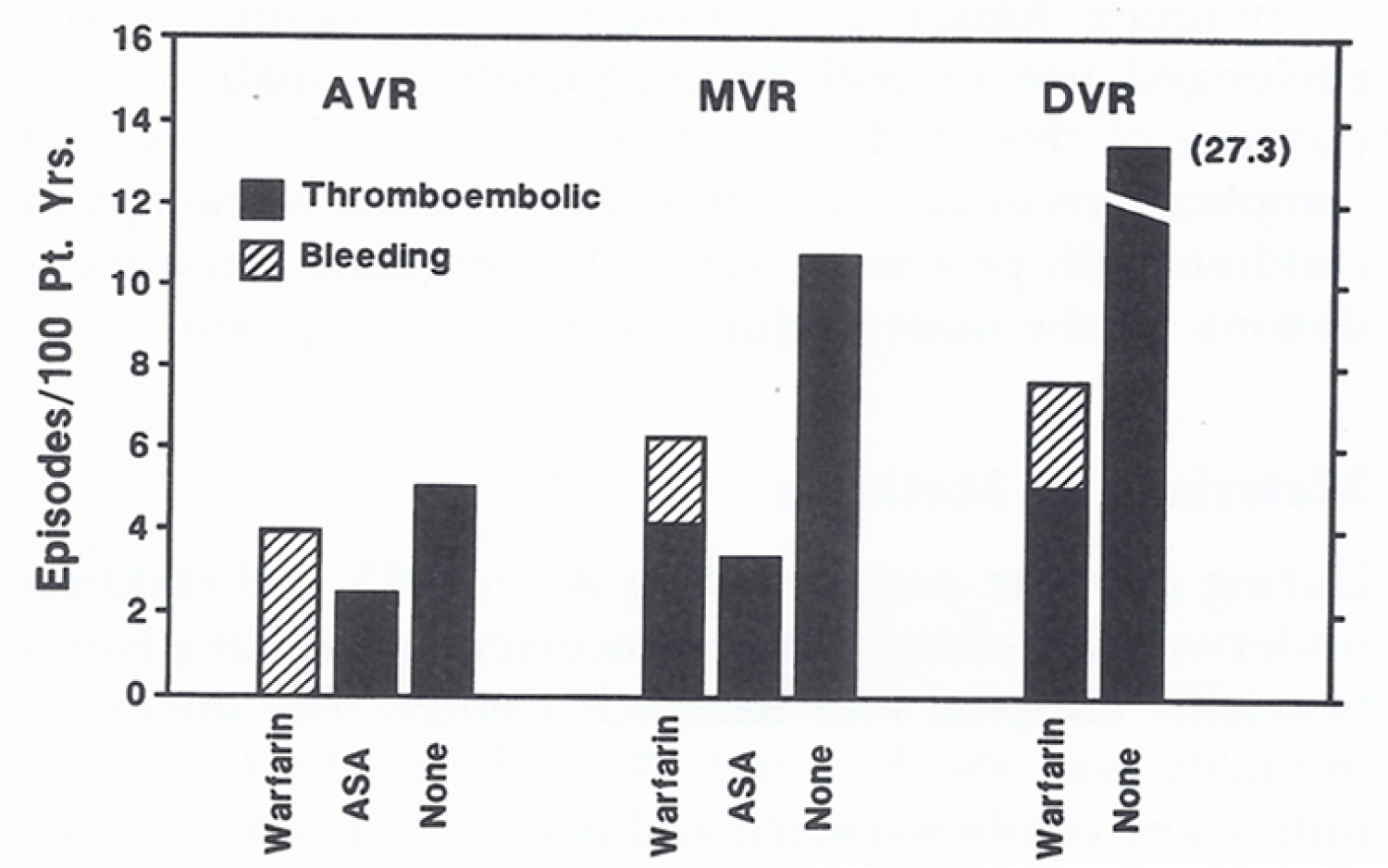 Figure 16: Thromboembolic and bleeding complications, expressed as episodes per 100 patient-years (Pt. Yrs.), based on anticoagulant treatment (warfarin sodium, aspirin plus dipyridamole [ASA], and no anticoagulants) and type of valve replacement (aortic valve [AVR], mitral valve [MVR], and double valve replacement [DVR]) [132].
View Figure 16
Figure 16: Thromboembolic and bleeding complications, expressed as episodes per 100 patient-years (Pt. Yrs.), based on anticoagulant treatment (warfarin sodium, aspirin plus dipyridamole [ASA], and no anticoagulants) and type of valve replacement (aortic valve [AVR], mitral valve [MVR], and double valve replacement [DVR]) [132].
View Figure 16
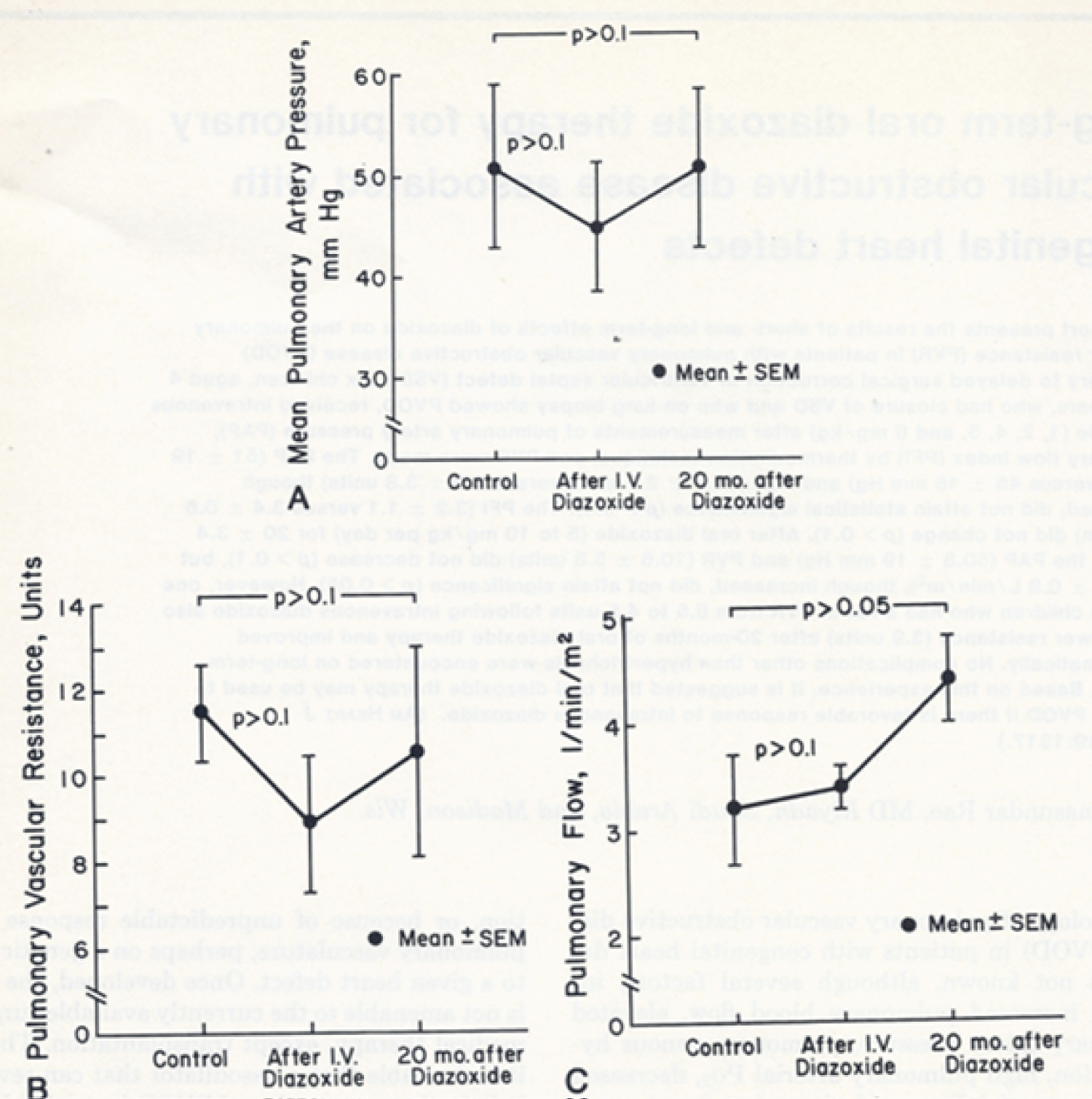 Figure 17: Response of pulmonary artery pressure (PAP), pulmonary vascular resistance (PVR), and pulmonary flow index to intravenous and long-term oral administration of diazoxide. There was a fall in the PAP (A) and PVR (B), but this did not attain statistical significance (p > 0.1) following intravenous administration of diazoxide (5 to 6 mg/kg). There was no significant (p > 0.1) change in the pulmonary flow index (C). After long-term (mean of 20 months) oral diazoxide, the PAP (A) and PVR (B) remained unchanged (p > 0.1). The pulmonary flow index increased (C), although this increase was not statistically significant (p > 0.05) [133].
View Figure 17
Figure 17: Response of pulmonary artery pressure (PAP), pulmonary vascular resistance (PVR), and pulmonary flow index to intravenous and long-term oral administration of diazoxide. There was a fall in the PAP (A) and PVR (B), but this did not attain statistical significance (p > 0.1) following intravenous administration of diazoxide (5 to 6 mg/kg). There was no significant (p > 0.1) change in the pulmonary flow index (C). After long-term (mean of 20 months) oral diazoxide, the PAP (A) and PVR (B) remained unchanged (p > 0.1). The pulmonary flow index increased (C), although this increase was not statistically significant (p > 0.05) [133].
View Figure 17
 Figure 18: The percent change in the pulmonary vascular resistance (PVR) immediately following intravenous diazoxide, and after long-term oral diazoxide administration, are shown. Note that there is no significant change in the PVR for the group as a whole (solid circles). In a single patient (open circle), the PVR fell following intravenous diazoxide and remained low after 20 months of oral diazoxide therapy [133].
View Figure 18
Figure 18: The percent change in the pulmonary vascular resistance (PVR) immediately following intravenous diazoxide, and after long-term oral diazoxide administration, are shown. Note that there is no significant change in the PVR for the group as a whole (solid circles). In a single patient (open circle), the PVR fell following intravenous diazoxide and remained low after 20 months of oral diazoxide therapy [133].
View Figure 18
Further review of perinatal circulatory physiology [133-140], prosthetic valves in children and adolescents [141], reassessment of usefulness of porcine heterografts in mitral location in children (Figure 19 and Figure 20) [142], cardiac murmurs in children [143], delayed presentation of anomalous circumflex coronary artery arising from pulmonary artery following prior surgical repair of aorto-pulmonary window in infancy [144], chest pain in children [145], congenital coronary artery anomalies [146], successful thrombolytic therapy of pulmonary embolism seen in association with urosepsis in an infant [147], current perspectives on Kawasaki disease [148], an approach to the diagnosis of cyanotic neonate for the primary care provider [111], principles of management of the neonate with CHD [110,140], cardiac emergencies in pediatric practice [149-152], development of supravalvar pulmonary artery stenosis following a Nuss procedure (Figure 21 and Figure 22) [153], consensus on timing of intervention for common congenital heart diseases [72,154], a review of childhood hypertension [155], prevention of sudden death in athletes [156] and state of the art management of CHDs [73,157] was presented.
 Figure 19: The actuarial valve survival curves for children ≤ 15 years are shown for mechanical and porcine heterografts; note the poor survival rate for heterografts (p = 0.015). The confidence limits are marked on only one side of the curve to clearly discern both the curves [143].
View Figure 19
Figure 19: The actuarial valve survival curves for children ≤ 15 years are shown for mechanical and porcine heterografts; note the poor survival rate for heterografts (p = 0.015). The confidence limits are marked on only one side of the curve to clearly discern both the curves [143].
View Figure 19
 Figure 20: The actuarial valve survival curves for children > 15 years are shown for mechanical and porcine heterografts; the survival curves are similar (p = 0.97). The confidence limits are marked on only one side of the curve to visualize both curves clearly [143].
View Figure 20
Figure 20: The actuarial valve survival curves for children > 15 years are shown for mechanical and porcine heterografts; the survival curves are similar (p = 0.97). The confidence limits are marked on only one side of the curve to visualize both curves clearly [143].
View Figure 20
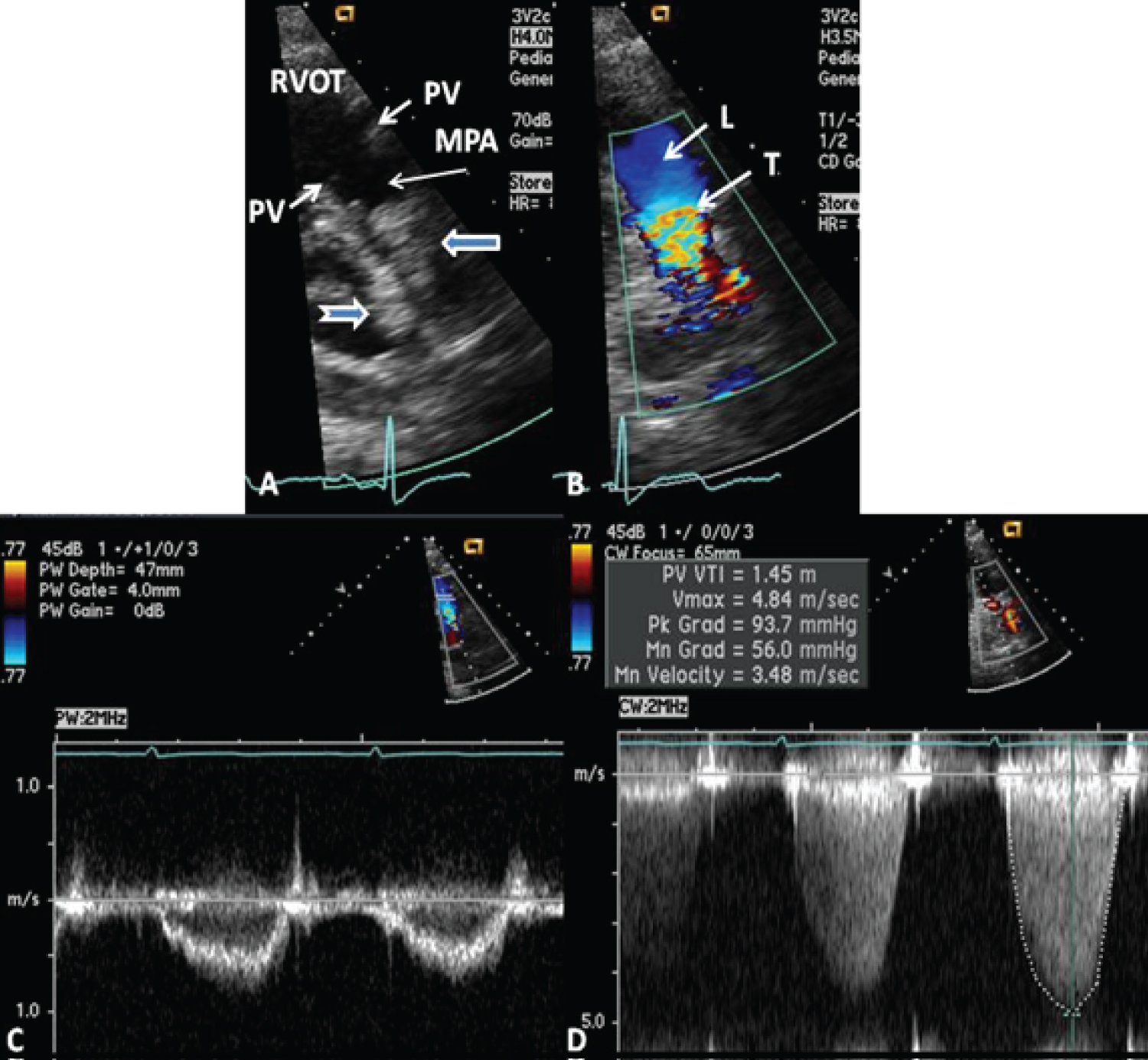 Figure 21: A) Selected video frame from a parasternal short axis view, showing echo-dense structures (thick blue arrows) within and outside the main pulmonary artery (MPA). The pulmonary valve (PV) leaflets (small arrows) are shown and appear normal. The right ventricular outflow tract (RVOT) and proximal MPA are free of any echo-dense structures; B) Color Doppler mapping of the same structures as in panel A shows normal laminar (L) flow in the RVOT and proximal MPA and turbulent (T) flow starting in the proximal MPA, indicating obstruction; C) Pulse Doppler sampling from the proximal MPA, which shows normal flow velocity; D) Continuous wave Doppler sampling demonstrating high velocity flow across the MPA, with a calculated peak instantaneous gradient of 93.7 mmHg and a mean gradient of 56 mmHg, indicating severe obstruction [154].
View Figure 21
Figure 21: A) Selected video frame from a parasternal short axis view, showing echo-dense structures (thick blue arrows) within and outside the main pulmonary artery (MPA). The pulmonary valve (PV) leaflets (small arrows) are shown and appear normal. The right ventricular outflow tract (RVOT) and proximal MPA are free of any echo-dense structures; B) Color Doppler mapping of the same structures as in panel A shows normal laminar (L) flow in the RVOT and proximal MPA and turbulent (T) flow starting in the proximal MPA, indicating obstruction; C) Pulse Doppler sampling from the proximal MPA, which shows normal flow velocity; D) Continuous wave Doppler sampling demonstrating high velocity flow across the MPA, with a calculated peak instantaneous gradient of 93.7 mmHg and a mean gradient of 56 mmHg, indicating severe obstruction [154].
View Figure 21
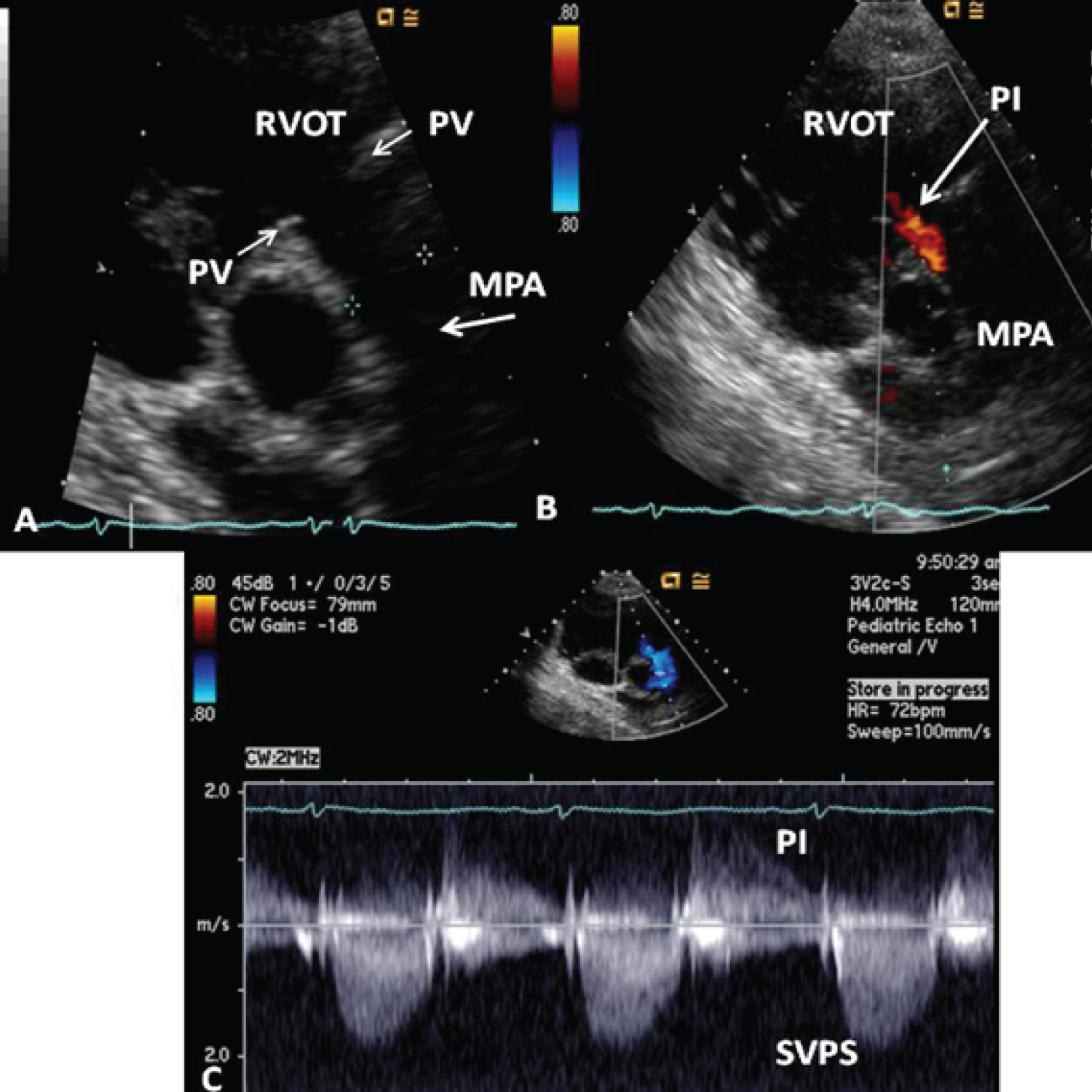 Figure 22: Echo-Doppler studies performed five months after the removal of the Nuss bar. A) Selected video frame from a parasternal short axis view, demonstrating the absence of the echo-dense structures in the right ventricular outflow tract (RVOT) and main pulmonary artery (MPA) that were seen prior to the removal of the Nuss bar (Figure 22-32A). The pulmonary valve (PV) leaflets (arrows) are shown; B) Color Doppler mapping of the same structures as in panel A, showing mild pulmonary insufficiency (PI) (arrow); C) Continuous wave Doppler sampling, demonstrating a low Doppler flow velocity across the MPA with a calculated peak instantaneous gradient of 15 mmHg, indicating minimal supravalvular pulmonary stenosis (SVPS) and mild pulmonary insufficiency (PI) [154].
View Figure 22
Figure 22: Echo-Doppler studies performed five months after the removal of the Nuss bar. A) Selected video frame from a parasternal short axis view, demonstrating the absence of the echo-dense structures in the right ventricular outflow tract (RVOT) and main pulmonary artery (MPA) that were seen prior to the removal of the Nuss bar (Figure 22-32A). The pulmonary valve (PV) leaflets (arrows) are shown; B) Color Doppler mapping of the same structures as in panel A, showing mild pulmonary insufficiency (PI) (arrow); C) Continuous wave Doppler sampling, demonstrating a low Doppler flow velocity across the MPA with a calculated peak instantaneous gradient of 15 mmHg, indicating minimal supravalvular pulmonary stenosis (SVPS) and mild pulmonary insufficiency (PI) [154].
View Figure 22
The subspecialty of Pediatric Cardiology was in early development when the author started his career in the mid-1960s. The author had the chance to observe first hand the stepwise evolution of Pediatric Cardiology over the last 50 years. Therefore, a unique historical perspective can be offered. The author has bestowed substantial interest in the development of new knowledge while providing care of patients with heart disease over a 50-year period. This book describes these developments along with the author's contributions to Pediatric Cardiology. The major focus of this book was to bring together the advances in Pediatric Cardiology, particularly the management of congenital heart defects. Along the way, the art and science of interventional pediatric cardiology were presented, as applicable to each chapter of the book. Developments such as early detection of the neonates with serious heart disease and their rapid transport to tertiary care centers, availability of highly sensitive noninvasive diagnostic tools, advances in neonatal care and anesthesia, progress in percutaneous interventional procedures and extension of complicated surgical procedures to the neonate and infant have advanced to such a degree that almost all congenital cardiac defects can be diagnosed and "corrected". The defects that could not be corrected could be effectively palliated. Cardiac defects that were once fatal in infancy are now treatable. These principles were incorporated into the respective chapters, as applicable [1]. Although every attempt was made to minimize repetition, there was some degree of replication which was unavoidable to preserve continuity of thought and discussion. The author hopes that the information presented is useful to the medical students, residents, fellows, other trainees, and paramedical personnel as well as to the pediatricians, neonatologists, pediatric intensivists, seasoned pediatric cardiologists, and pediatric cardiovascular surgeons [1].