Coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 or SARS-CoV-2 has been declared a pandemic. Broadly, hospitalized COVID-19 pneumonia patients suffer two important cardiac effects. Firstly, when COVID-19 pneumonia affects people with pre-existing cardiovascular disease (CVD), they have worst outcomes compared to those without CVD. Secondly, COVID-19 per se causes myocardial injury, myocarditis, myopericarditis, arrhythmias, ventricular dysfunction and heart failure. As many manifestations of COVID-19 sepsis mimics primary cardiac illness, physicians and intensivists managing COVID-19 are in a dilemma with regard to presence or absence of primary cardiac involvement in a COVID-19 pneumonia patient. Hence, it is prudent for all physicians and cardiologists to be aware about the diagnosis and management of COVID-19 related cardiac illness for proper management of the patient. In this review, we summarize all the available literature and guidelines on how COVID-19 pneumonia affected hospitalized patients with pre-existing CVD have worst outcomes and the possible mechanisms behind it as well as COVID-19 de-novo cardiac manifestations and their management.
COVID-19, SARS-CoV-2, Pneumonia, Cardiovascular diseases, Myocardial injury, Myocarditis, Troponin, Echocardiography, Acute coronary syndrome, Myocardial infarction, Percutaneous coronary intervention
Coronavirus disease 2019 (COVID-19) patients with pre-existing cardiovascular disease (CVD) have been found to be at high risk for adverse events and infection per-se is associated with acute cardiac injury [1-4]. Previous studies have noted myocardial injury, myocarditis, myopericarditis, cardiac arrhythmias, ventricular systolic dysfunction, and heart failure (HF) in patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections [5-7]. As many manifestations of COVID-19 sepsis mimics primary cardiac illness, physicians and intensivists managing COVID-19 are in a dilemma with regard to presence or absence of primary cardiac involvement in a COVID-19 pneumonia patient. In this review, we summarize how COVID-19 pneumonia affected hospitalized patients (either in COVID-19 ambulatory ward or intensive care unit [ICU]) with pre-existing CVD have worst outcomes and the possible mechanisms behind it as well as COVID-19 de-novo in-hospital cardiac manifestations and their management. Those patients who primarily present with cardiac manifestations e.g. ST-elevation myocardial infarction (STEMI) then tested positive for SARS-CoV-2 as well as non-hospitalized SARS-CoV-2 positive patients are excluded from this review.
Prior studies demonstrated that SARS-CoV-2 primarily transmitted by the respiratory route binds its viral spike (S) proteins to angiotensin-converting enzyme 2 (ACE2) receptor proteins for cell entry [8,9]. The virus shows strong binding to soluble ACE2 receptors and cell-associated entry into several organs such as the kidneys, lung, heart, brain, liver, and intestine [10]. After gaining entry into the cells via ACE2 receptors, SARS-CoV-2 down-regulates the expression of ACE2 so that the enzyme is unable to exert protective effects on various organs [11]. ACE2 is highly expressed in alveolar lung cells, vascular endothelium, heart and plays a role in the lung and cardiac damage; thus, viral binding to this receptor induce endothelial inflammation in various organs due to both viral infection and the host’s own immune response, which contributes to viral pathogenicity [12,13].
It has been demonstrated that there is presence of viral elements within endothelial cells along with inflammatory cells, with evidence of endothelial and inflammatory cell death leading to diffuse endotheliitis in several organs [14]. In addition, induction of apoptosis and pyroptosis leads to endothelial cell injury in patients with COVID-19 which can explain the systemic impaired microcirculatory function in different vascular beds [14]. Furthermore, myocardial injury is demonstrated in Covid-19 patients who may be due to direct endothelial or vascular injury, hypoxic injury, cytokine storm, coronary spasm, and a procoagulant state with micro thrombi [13-16]. Cardiac magnetic resonance imaging (MRI) has shown myocardial interstitial edema [17].
Different studies from different regions have reported varying prevalence of CVD among COVID-19 affected patients. It has been noted that COVID-19 patients with CVD have worst prognosis compared to those without CVD. In the first study of 138 COVID-19 hospitalized patients from China, cardiovascular co-morbidities were 46% and among them 72% needed intensive care unit (ICU) care. In addition among 15% CVD patients, 25% of them required ICU care [1]. This earlier study showed very high prevalence of CVD and its risk factors indicating initial involvement of vulnerable population with significant CVD.
Later as the infection spread across all population, the prevalence of CVD among COVID-19 patients came down, but was still high among sicker patients. In a study involving 1,099 confirmed COVID-19 patients from China, 15.74% had a serious illness, with 23.7% hypertensives, 5.8% had ischemic heart disease (IHD), and 2.3% had a cerebrovascular disorder [18]. In another study, results from 22,512 COVID-19 patients in the Italian population found that 7.2% patients died in the cohort. A subsample of 350 ICU patients showed, 30% had an IHD, 24.5% had atrial fibrillation (AF), and 9.6% had history of stroke [19]. A meta-analysis of eight reports from China involving 46,248 COVID-19 patients revealed that most common co-morbidities were hypertension (17 ± 7, 95% CI 14-22%), diabetes (8 ± 6, 95% CI 6-11%) followed by CVD (5 ± 4, 95% CI 4-7%) [20]. Data from China's National Health Commission found that 35% of COVID-19-diagnosed patients were hypertensive and 17% had CVD [13].
In an analysis of 44,672 confirmed COVID-19 cases from Wuhan, China, there was high case fatality rates (CFR) in patients with CVD (10.5%), diabetes (7.3%), and hypertensives (6.0%); all markedly higher than the 2.3% overall CFR [21]. In a meta-analysis, a total of 18 studies (n = 4858 patients) were included. Pre-existing CVD was associated with a significantly increased risk of a severe form of COVID-19 (OR = 3.14) and overall risk of COVID-19 all-cause mortality (OR = 11.08) [4]. Initially, it was understood that elderly population are known to have more CVD and hence they were prone for severe infection and adverse events. However, in another meta-analysis, included 51 studies with a total of 48,317 patients with confirmed COVID-19 infection, overall, the relative risk of developing severe COVID-19 or death was significantly higher in patients with hypertension (OR 2.50), diabetes (OR 2.25) and CVD (OR 3.11) across all ages [22]. Interestingly, though young patients had lower prevalence rates of CVD than elderly patients, relative risk of fatal outcome in young patients with hypertension, diabetes and CVD was higher than in elderly patients. Thus, underlying CVD patients have worse prognosis compared to those without CVD irrespective of age.
At present, many mechanisms are hypothesized for this association of severity of COVID-19 and pre-existing CVD. Possible explanations include, CVD patients developing ‘diffuse endotheliitis’ as mentioned before, a functionally impaired immune system precipitating prothrombotic antibodies, hypoxia-induced myocardial injury in already compromised cardiac condition, small vessel ischemia due to microvascular injury and thrombosis [23,24]. In addition, majority of CVD patients are on angiotensin-converting enzyme 1 inhibitors (ACEI)/angiotensin receptor blockers (ARB) and it’s hypothesized that it may reciprocally increase ACE2 expression and risk of severe SARS-CoV-2 infection. However, large observational studies reported that virus internalization via ACE2 may cause ACE2 depletion (which normally metabolizes angiotensin II) that may lead to high angiotensin II activity which is countered by ACEI and ARBs [3,24]. Hence, all major societies have recommended against adding or stopping ACEI/ARBs in COVID-19 patients unless indicated [25].
Even though racial and ethnic disparities were observed in earlier studies, a large analysis of the American Heart Association (AHA), COVID-19 Cardiovascular Disease Registry of 7868 patients hospitalized with COVID-19 did not show any disparity [26]. It is observed that only way to prevent these adverse effects of COVID-19 on patients with known CVD is by controlling of cardiac risk factors, vaccination, as well as providing good hospital and ICU care according to guideline recommended treatments for COVID-19 infection [24-26].
Cardiac manifestations of COVID-19 can be broadly categorized as (1) indirect and (2) direct cardiac insults [1,6,7,27-34]. Indirect cardiac manifestations noted in patients with COVID-19 are myocardial injury, right ventricular (RV) dysfunction, septic cardiomyopathy, stress cardiomyopathy and heart failure with preserved ejection fraction (HFpEF). Direct cardiac manifestations are myocarditis with ventricular dysfunction and heart failure with reduced ejection fraction (HFrEF), myopericarditis with or without effusion, cardiac arrhythmias and cardiogenic shock [1,6,7,27-34]. It can precipitate acute coronary syndrome (ACS) secondary to viral-mediated coagulopathy and extensive thrombosis [16]. The common difficulty faced by physicians in daily clinical practice is to differentiate myocardial injury vs. myocarditis vs. ACS. Myocardial injury can be classified into two types, with or without echocardiographic abnormalities [31].
The term “myocardial injury” applies when at least one cardiac troponin (cTn) concentration is above the 99th percentile upper reference limit (URL) [27,34]. SARS-CoV-2 has the potential to cause myocardial damage evidenced by elevated cardiac biomarkers. Prevalence of myocardial injury is variable, generally between 20 to 30% [24,31]. In the samples of 138 hospitalized patients in Wuhan provenience of China, 7.2% overall patients and 22% of ICU based patient developed very high levels of cTn I [1]. In 416 hospitalized patients with COVID-19, 19.7% had myocardial injury. Compared with patients without cardiac injury, those with elevated high-sensitivity cTn I (median [IQR], 0.19 [0.08-1.12] vs. < 0.006 [< 0.006-0.009] μg/L) and elevated N-terminal pro-B-type natriuretic peptide (median [IQR], 1689 [698-3327] vs. 139 [51-335] pg/mL) required increased noninvasive or invasive mechanical ventilation [6].
Patients with myocardial injury had higher mortality than those without myocardial injury (42 of 82 [51.2%] vs. 15 of 334 [4.5%]; P < .001) [6]. In a meta-analysis of 1527, COVID-19 patients in six studies revealed that acute myocardial injury was seen in 8% of total patients [7]. The incidence of acute myocardial injury was about 13 folds higher in ICU/severe pneumonia patients compared with the non-ICU/severe patients [7]. These increases in cardiac biomarkers are more likely to occur in those with severe COVID-19 presentations. It’s noted that, there is progressive increase in cTn and natriuretic peptides (NP) levels during hospitalization in non survivors, but not among survivors [2,3].
The increase in cardiac biomarkers may be due to three potential causes [27,34]. Firstly, chronic myocardial injury (Chronic HF, cardiomyopathy, chronic kidney disease) may present with pre-existing raised baseline cTn; Secondly, it may represent acute nonischemic myocardial injury secondary to direct cardiac insult (SARS-CoV-2 myocarditis, pericarditis, arrhythmias, ventricular dysfunction with HFrEF) or due to indirect cardiac insult (RV strain due to lung disease or pulmonary embolism, sepsis, septic cardiomyopathy, cytokine storm) [25,27,34]. They may or may not have any electrocardiogram (ECG) changes. They may present with ST elevation on ECG, without regional wall motion abnormalities on echocardiogram and with normal coronaries [17,25,35]. Thirdly, it could represent ACS either due to type 1 MI (plaque rupture with thrombus formation) or due to type 2 MI (hypoxia, spasm, arrhythmias, hypertension or hypotension) [25,27,34,35].
So, the predominant causes of myocardial injury leading to high Troponin release with normal echocardiogram are indirect insults to heart from COPVID-19 infection, namely, right ventricular strain due to lung disease or pulmonary embolism, sepsis, cytokine storm with ventricular dysfunction and the causes of type 2 MI i.e., hypoxia, coronary spasm, arrhythmias, hypertension or hypotension.
It is very important to identify myocardial injury vs. ACS, as based on cTn raise alone doing a high risk invasive procedure in Cath lab may expose health care workers for infection [5,27,34]. Wang, et al. in their study reported that 41% of their cases were healthcare-associated [1]. In a study from Oman, among COVID-19 positive health care workers, 25% were hospital acquired [35]. Two important observations in differentiating myocardial injury from ACS are: cTn concentrations in myocardial injury are often lower than seen in acute myocardial infarction (MI) and that if cTn proportionally rises with other inflammatory biomarkers (D-dimer, ferritin, lactate dehydrogenase and interleukin-6) it reflects cytokine storm more than ACS [25,34].
Sandoval, et al. opine that not everyone with cTn increases > 99th percentile (or even with an increasing pattern) requires imaging like echocardiography or computed tomography (CT) scan [34]. If the increases are mild to moderate and stable on repeating with normal ECG, imaging may not be needed. Selective imaging can be considered with very marked cTn increases like seen in acute MI or myocarditis [34]. Echocardiography should not routinely be performed in patients with COVID-19 disease [36]. According to the EACVI 2020 recommendations, cardiac MRI to diagnose myocarditis or CT coronary angiogram to diagnose ACS should only be performed if the expected information will change management as this involves risk of transportation of critically ill patients who may deteriorate during transportation, risk of infection for professionals (technicians, nurses, medical orderlies), and risk of contamination of equipment and facilities [36].
Based on available literature discussed above, we suggest doing ECG in all COVID-19 admitted patients with high cTn levels. If ECG shows no ST-T changes, management of “myocardial injury” is observation with serial ECGs and cTn performed daily once i.e. every 24 hours. A downward trend suggests myocardial injury. If ECG shows significant ST-T changes and cTn very high, a bedside echocardiogram has to be done to see for chamber dilatation, global and regional wall motion abnormalities, wall thickness, ejection fraction (EF), RV function (TAPSE/FAC) and RV systolic pressure i.e. a focused cardiac ultrasound study (FoCUS) must be performed in short time period without connecting ECG leads to the patient [36]. If echocardiogram is normal (commonly in myocardial injury its normal), a diagnosis of “myocardial injury” may be considered and followed up medically. Only exception is presence of RV dilatation and dysfunction and its presence is associated with severe lung disease (acute respiratory distress syndrome) or pulmonary embolism which should be investigated with a CT pulmonary angiogram and managed accordingly depending on clinical condition of the patient [37]. In contrast, LV size is generally normal and LV function is hyperdynamic in most, whereas significant valvular abnormalities, either primary or secondary, are absent [37].
cTn raise is common in myocarditis, but differentiation between “myocardial injury” and “myocarditis” is challenging. Endomyocardial biopsy (EMB) is the gold-standard for diagnosis of myocarditis, which is not practical in a sick COVID-19 patient, lack of expertise, false negatives and non-availability in majority of hospitals. In the absence of EMB, a diagnosis of myocarditis can be made from clinical, ECG, biochemical, echocardiographic study and cardiac MRI showing evidence of regional or global high T2 signal, early myocardial enhancement and/or patchy non-ischemic late gadolinium enhancement, myocardial edema and/or scarring. The occurrence of left ventricular dysfunction or pericardial effusion provides additional supportive evidence for myocarditis [17,38]. The pathophysiology of COVID-19-related myocarditis is thought to be a combination of direct viral replication in cardiac myocytes and cardiac damage due to the host’s T-lymphocyte mediated cytokine immune response [24,39].
In a study of Covid-19 positive patients with raised high-sensitivity cTn who underwent cardiac MRI, overall a diagnosis of myocarditis was noted in 1-2% of the total number of SARS-CoV-2 positive patients, with pericardial effusion seen in approximately 10% of patients with COVID-19 myocarditis [40]. Furthermore, in German cardiac MRI study, among 100 patients recovered from COVID-19, 60% of patients had evidence of myocardial inflammation by MRI, but only 5% had a cTn raised above the reference range (> 14 pg/mL) [41].
Diagnosing COVID-19-related “myocarditis” is difficult as in any other myocarditis cases, more so in COVID-19 positive patient. Patients may complain of recurrent chest pain or chest tightness. Severe myocarditis may present with acute HF (HFrEF) or cardiogenic shock as in fulminant myocarditis which will be difficult to differentiate clinically from COVID-19 related septic shock or septic cardiomyopathy which is usually reversible [39]. cTn and NPs usually elevated as in myocardial injury, but at higher levels due to possible ventricular dilation and wall edema [39]. Various ECG abnormalities are seen in myocarditis ranging from ST elevation (Figure 1), PR depression, new-onset bundle branch block, QT prolongation, pseudo infarct pattern, premature ventricular complexes, and tachy or bradyarrhythmias with advanced atrioventricular nodal block [38]. The American Heart Association (AHA) recommends 1 or more cardiac imaging methods such as echocardiogram or cardiac MRI in patients with myocarditis [42]. The cardinal signs of myocarditis on echocardiogram are increased wall thickness due to edema, chamber dilation, pericardial effusion, regional or global wall motion abnormalities with ventricular systolic dysfunction, pericardial thickening or effusion which differentiates it from myocardial injury [38,42,43] (Video 1). In a meta-analysis, approximately 5% of patients with COVID-19 who underwent CT chest had pericardial effusion [44]. The pericardial fluid is generally sterile indicating an inflammatory response rather than direct infection [38].
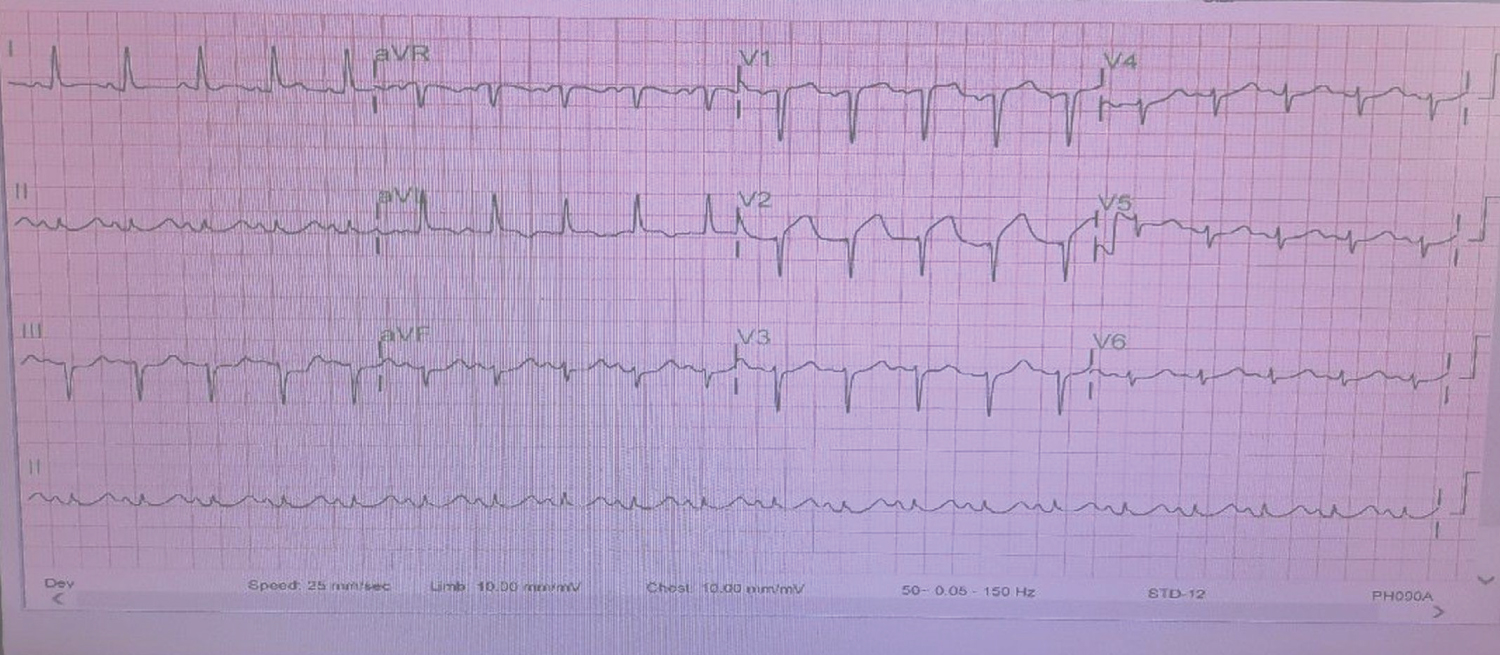 Figure 1: ECG of a 64 year women with Covid-19 pneumonia with chest pain showing ST elevation anterior leads with Troponin rising from 43 pg/ml to 756 pg/ml.
View Figure 1
Figure 1: ECG of a 64 year women with Covid-19 pneumonia with chest pain showing ST elevation anterior leads with Troponin rising from 43 pg/ml to 756 pg/ml.
View Figure 1
Video 1: Echocardiogram of same patient in Figure 1 showing severe regional wall motion abnormalities resulting in akinesia of apex, mid to apical segments of septum and anterolateral walls with severe LV systolic dysfunction.
Differentiating myocarditis from ACS is again challenging. There are no published management guidelines for admitted COVID-19 patients who develop ACS. If echocardiogram is abnormal it may indicate myocarditis or ischemic etiology as regional wall motion abnormalities seen in both. If there is no ST elevation, but significant ST-T changes, very high cTn, multiple cardiac risk factors and high suspicion of non-ST elevation MI (NONSTEMI) then patient should be treated medically with antiplatelets, high-dose statins, beta blockers and therapeutic LMWH (48 hours) as in usual NONSTEMI (ACC/SCAI [45], ESC[46]). A CT coronary angiogram can be done (EACVI[36], EAPCI [46]) and if it shows severe lesions or patient has recurrent chest pain, dynamic ECG changes and/or “new” hemodynamic instability then to perform urgent percutaneous coronary intervention (PCI) [27,45-47]. If CT coronary angiogram not done, if stable patient can be brought for elective PCI after 4 weeks. If CT coronary angiogram done and is normal or non-obstructive coronary artery disease, then a diagnosis of “myocarditis” can be presumed and if possible check for CT signs of “myocarditis” during same study or do cardiac MRI for myocarditis confirmation [36,46]. However, as there is no specific treatment for myocarditis, we propose that cardiac MRI can be avoided if CT coronary angiogram is normal. ACEI/ARB should not be started unless indicated.
Most guidelines recommendations are for COVID-19 patients presenting as STEMI which can to some extent be extrapolated to in-hospital STEMI occurrence in COVID-19 pneumonia admitted patients. The joint statement from the ACC Interventional Council and SCAI said that Fibrinolysis can be considered an option for a relatively stable patient with STEMI with active COVID-19 [45]. The EAPCI Position Statement on Invasive Management of Acute Coronary Syndromes during the COVID-19 pandemic states that decisions should be made (primary PCI or thrombolysis) taking into account the potential extra delays in target times [46]. Primary PCI is first-line therapy if it can be performed within 120 min from symptom onset including time to put on PPE. Fibrinolysis if not contraindicated can be considered when the delay is longer. Complete revascularization to be considered if indicated and appropriate. Left ventricular angiogram instead of echocardiogram to evaluate left ventricular function.
According to Chinese Society of Cardiology recommendations, all STEMI should be thrombolysed as first-line treatment and advised further conservative treatment along with patients of STEMI late presenters and stable high risk NONSTEMI [47]. Conditions warranting invasive intervention are limited to STEMI with hemodynamic instability and life-threatening NONSTEMI [47]. In addition they specify that interventions should be done in a cath lab with negative-pressure ventilation, strict high quality PPE for staff and with strict periprocedural disinfection.
The American SCAI Emerging Leader Mentorship Members and Graduates group recommends that a subset of patients should be considered for deferral of urgent diagnostic or interventional procedures in the catheterization laboratory during the pandemic: STEMI patients with severe pneumonia should undergo conservative treatment which include fibrinolytic therapy and NONSTEMI with low-risk features stabilized on medical therapy (i.e. without refractory chest pain, or evidence of hemodynamic or electrical instability) [48,49]. Furthermore, cardiologists should recognize that COVID-19 patients with STEMI during cytokine storm have extensive macrovascular/microvascular thrombosis, myocardial edema which may result in poor coronary flow and worsening outcomes [16]. In a large STEMI registry of COVID-19 patients, 1,185 patients were included in the NACMI registry (230 COVID positive patients, 495 suspected COVID, and 460 non-COVID). Among COVID positive patients who underwent coronary angiogram, 71% received primary PCI and 20% received medical therapy. The primary outcome occurred in 36% of COVID positive patients, 13% of suspected Covid and 5% of control patients. So 1 in 5 had ST elevation due to other causes and 1 in 3 expired due to COVID illness in spite of Primary PCI.
Considering above recommendations we suggest, for typical in-hospital STEMI on ECG, first-line treatment should be thrombolysis (as the main aim of initial revascularization is restoration of coronary flow). If patient has unsuccessful thrombolysis or hemodynamically unstable then patient needs to be taken for rescue PCI taking into consideration severity of COVID pneumonia, sepsis, ventilation, multiorgan involvement and probability of any recovery from COVID pneumonia. If patient has successful thrombolysis, patient can undergo elective PCI after 4 weeks once treated for pneumonia. If taken for primary PCI and coronary angiogram turns out to be negative for significant coronary artery disease, a diagnosis of myocarditis can be considered as cause for ST elevation (Video 2).
Video 2: Coronary angiogram of same patient in Figure 1 showing normal coronary anatomy suggestive of Covid-19 myocarditis as overall presentation.
Management of COVID-19 myocarditis is supportive including mechanical circulatory support if available and depending upon seriousness of lung problem [5,27]. When HFrEF occurs ACEI, Beta blockers and aldosterone antagonists have to be started. Few case reports of intravenous immunoglobulin and corticosteroids use in COVID-19 myocarditis are reported, but not endorsed by any guidelines [43]. In a study, rates of in-hospital mortality were 5.2% in patients without myocardial injury, 18.6% with myocardial injury without echocardiographic abnormalities and 31.7% with myocardial injury and echocardiographic abnormalities which indicates myocarditis has bad prognosis among the three groups [31].
Earlier reported COVID-19 studies in 138 hospitalized patients recorded 17% cases of cardiac arrhythmia, 44% of ICU patients [1]. Guo, et al. reported a 5.9% (11/187) incidence of ventricular tachycardia (VT)/ventricular fibrillation (VF) in COVID-19 patients [2]. Common arrhythmias reported in COVID-19 patients are sinus tachycardia, sinus bradycardia, atrial fibrillation, atrial flutter, polymorphic ventricular tachycardia, high grade atrio-ventricular (AV) block [19,50-55]. In a large Swedish cardiac arrest registry, COVID-19 was responsible in 16% of in-hospital cardiac arrest, VT/VF seen in 18.5% and 30-day mortality was increased 2.3-fold in these patients [55]. In a cohort of 121 COVID-19 patients, Yu, et al. demonstrated that sinus tachycardia was the commonest arrhythmia at 72% [50]. Persistent tachycardia mean duration was 12.7 days with a mean heart rate of 117 beats/min (range: 102‐150 beats/min) and in 40% of patients it persisted at 30 days (50). Transient sinus bradycardia was seen in 18 (14.9%) patients with a mean heart rate of 43 beats/min (range: 38‐49 beats/min) and a mean duration of 2.6 days (50). In another study of 115 patients atrial tachyarrhythmia’s was seen in 16.5% patients, among all admitted to the ICU (27.5%); which included AF in 12 patients, atrial flutter in 6 patients, and atrial tachycardia in 1 patient [51].
Atrial or ventricular arrhythmias in an ICU patient are multifactorial and generally result from electrolyte abnormalities, hypoxia, and acidosis and catecholaminergic release as in any patient with sepsis [52-54]. However, if the above causes are ruled out then the development of cardiac arrhythmias, especially in the setting of elevated cardiac biomarkers in a COVID-19 patient indicate development of SARS-CoV-2 “myocarditis” [39,52,53]. The possible pathophysiology mechanisms of COVID-19 arrhythmias are direct injury to cardiomyocytes disrupting the plasma membrane and electrical conduction, microvascular ischemia, proinflammatory cytokines, myocardial fibrosis or scars causing re-entrant arrhythmias and extreme anxiety leading to further endogenous catecholamine release, causing myocardial electrical instability [39,54,55].
Managing arrhythmias is crucial in already compromised patient to reduce mortality and morbidity. New onset AF in patients with COVID-19 has been associated with poor outcomes in critically ill patients [19]. Symptomatic AV blocks with low heart rate may require temporary transvenous pacing if available and logistically possible to insert at bedside with fluoroscopy guidance or dopamine infusion may be administered instead of isoproterenol to avoid inadvertent β-2 receptor agonism [50]. For patients with sinus tachycardia, Ivabradine or diltiazem may be used for rate control [56]. Diltiazem or verapamil were also considered first in patients with atrial premature beats or supraventricular tachycardia without cardiac disease [56]. Beta-blockers should be used with caution due to bronchospasm in patients with COVID-19. Tachyarrhythmias may respond to antiarrhythmic drugs amiodarone or lidocaine/mexiletine which do not prolong QT interval or DC Cardioversion [53,54]. Sustained VT or AF can be treated with amiodarone if QT interval below 500 or DC Cardiversion [56]. In patients with baseline prolonged QTc intervals, particularly > 500 ms, the risks of QT prolonging drugs which are generally given for COVID-19 treatment causing polymorphic VT may outweigh the benefits and should be avoided [53,54].
Earlier studies from china reported about 4% prevalence of HF in hospitalized COVID-19 patients [2,6]. In a study of 3080 confirmed COVID-19 infection patients, prevalence of history of chronic HF was 4.9%, acute decompensated HF was seen in 11.2% of these patients and mortality was high among them when compared to those with no HF (48.7% vs. 19.0%; P < 0.001). In contrast, 2.5% were diagnosed with new onset AHF, and they also had higher mortality (46.8% vs. 19.7%; P < 0.001) [57]. This supports the potential of SARS-CoV-2 to induce “myocardial injury or myocarditis” due to various mechanism described above leading to either acute decompensation of chronic HF or new-onset AHF [2,57].
The overlapping clinical, ECG, biochemical and radiological presentations of both COVID-19 and HF make it difficult in accurately diagnosing HF [36,58,59]. Presence of pedal edema, JVP elevation, S3 gallop, chest X-ray showing alveolar edema mainly in lung bases with cardiomegaly in patients with HF whereas diffuse interstitial and alveolar lung edema in COVID-19 pneumonia and on chest CT predominantly central and basal lung opacities, basal pleural effusions, right side more than left with cardiac enlargement and rapid response to diuretic therapy seen in HF whereas bilateral consolidations, peripheral, diffuse ground-glass opacities, reticular patterns, air bronchograms, noted in COVID-19 pneumonia [58,59]. In addition, disproportionately high values of NPs seen in HF patients as well as low values has a high negative predictive value and may exclude HF in COVID-19 patients [57-59].
Echocardiography must be considered in all patients with suspected HF in hospitalized COVID-19 patients to evaluate right and left side of heart as described in the above discussions. In addition, inferior vena cava diameter and respiratory variation by echocardiography may be used to assess fluid status [58]. In an echocardiographic study of 100 patients with COVID-19, left ventricular EF was normal in 90% (HFpEF), and the most common abnormalities were right ventricular dilation (39%) and left ventricular diastolic dysfunction (16%) [28,32,60]. The mechanism for HFpEF in COVID-19 are same described in “myocardial injury” and those with HFrEF are possibly due to myocarditis as MRI studies have shown high incidence of myocarditis features in COVID-19 survivors [28,41].
Guideline-directed HF medical therapy (including beta-blocker, ACEi/ARB or ARNI, mineralocorticoid receptor antagonist, Ivabradine) along with diuretics must be started when indicated or continued if already on, unless BP is low, when their dosage can be reduced or discontinued [58]. At present, among the beta blockers approved for HF, experimental studies have shown that carvedilol preferable in COVID-19 patients due to its anti-cytokine properties [61].
Hospitalized patients with COVID-19 pneumonia have worse outcomes in presence of prior CVD risk factors and conditions. They are prone for decompensation needing prolonged care including ICU admission and ventilation. This calls for urgent need to make aware among the physicians and community for strict control and treatment of prior CVD conditions and encourage patients to get vaccinated. Those with severe co-morbid CVD conditions diagnosed with COVID-19 pneumonia have to be managed in a higher center for better prognosis as mortality and morbidity is high in these patients. In addition, COVID-19 per-se causes myocardial injury, myocarditis and precipitates ACS. To diagnose myocardial injury serial ECGs and cTn need to be done every 24 hours along with an echocardiogram if cTn very high with increasing trend. To differentiate possible myocarditis and NONSTEMI a CT coronary angiogram would suffice after starting treatment for NONSTEMI. Cardiac MRI in sick patient not advisable to confirm myocarditis as management will not change. Typical STEMI on ECG in hospitalized COVID-19 pneumonia patients should undergo fibrinolysis as treatment option and then considered for rescue PCI if indicated considering seriousness of COVID-19 pneumonia and patient overall condition. All ACS diagnosed patients if no in-hospital PCI done can be brought after 4 weeks or later for elective PCI once they recover from their COVID illness. Heart failure and cardiac arrhythmias occurring during COVID-19 pneumonia need to be managed in usual manner avoiding drugs causing QT prolongation whenever possible.
None.