Elevated level of lipoprotein (a) gene is the main cause of inherited risk of myocardial infarction (MI). Several RNA based therapies are being developed to reduce the expression of LP(a) gene, but none of them are neither available in market, nor have been declared as perfect choice for treatment. An American multinational biopharmaceutical company, Amgen Inc, has recently disclosed remarkable test results of phase II trial of Olpasiran [a small interfering RNA-based (siRNA) therapy to target LP(a) gene]. In this article, an overview has been provided to point up the potential risk of elevated LP(a) levels resulting in premature MI. Relevant resources were identified and abstracted by conducting a standard database research to summarize the approach of siRNA-induced gene silencing and identifying the most effective choice of treatment for lowering LP(a) levels, in this emerging era of related RNA therapeutics. Overviewing the siRNA strategy of gene silencing and comparing its effectiveness with other known therapies has shown that siRNA therapy is the most efficient treatment for lowering LP(a) levels and can be the future of gene therapies for cardiovascular diseases (CVDs).
Myocardial infarction, Lipoprotein(a), Small interfering RNA (siRNA), Cardiovascular diseases (CVDs)
According to world health organization (WHO), the first and foremost leading cause of worldwide deaths is heart disease, representing more than 16% of the total deaths for the last 20 years and the number of deaths has been increased from 2 to 9 million since 2000 to 2019 [1]. It was estimated in 2011 that the prevalence of cardiovascular diseases (CVDs) will reach to 40% in 2030 [2]. This prediction was surpassed in 2015, and now it is estimated that around 45% population of US will be suffered from either preclinical risks of CVDs or having high risk of sudden heart failures [2]. It has also been estimated that the global cost of tackling CVDs (either directly or indirectly) is $863 billion in 2010 which is expected to reach more than $1 trillion in 2030 [3]. Myocardial infarction (MI) also known as heart attack, is one of the most common heart diseases having complex etiology that involves both environmental as well as genetic risk factor or susceptibility. Due to recent advancements in genetic studies, it has been estimated that around 40-50% of MI as well as acute myocardial infarction (AMI) risk is based on hereditary factors that mostly includes young patients having low or even no clinical risk factor of MI [4]. Several RNA based therapies including messenger RNA (mRNA), microRNA (miRNA) and small interfering RNA (siRNA) has been discovered recently to treat the genetic risk of CVDs including MI, but the recent trial of siRNA-based therapy has shown more promising and long-lasting results of treatment. For example, Amgen's Olpasiran (also known as AMG 890) which is first-in-class N-acetylglucosamine (GalNAc)-conjugated siRNA-based therapy has shown potential effects on reducing the genetic risk of MI by inhibiting Lipoprotein(a) [LP(a)] mRNA translation and reducing more than 80% of LP(a) levels [5]. During the phase 1 trial of siRNA based Opasiran therapy, 71-90% of LP(a) levels decreased in the participants by receiving less than 9 mg dose. While in phase 2 trial that has recently been disclosed by Amgen, siRNA based Opasiran therapies are believed to have slashed more than 90% of LP(a) levels but the clinical results are yet to be declared [5,6] (Figure 1).
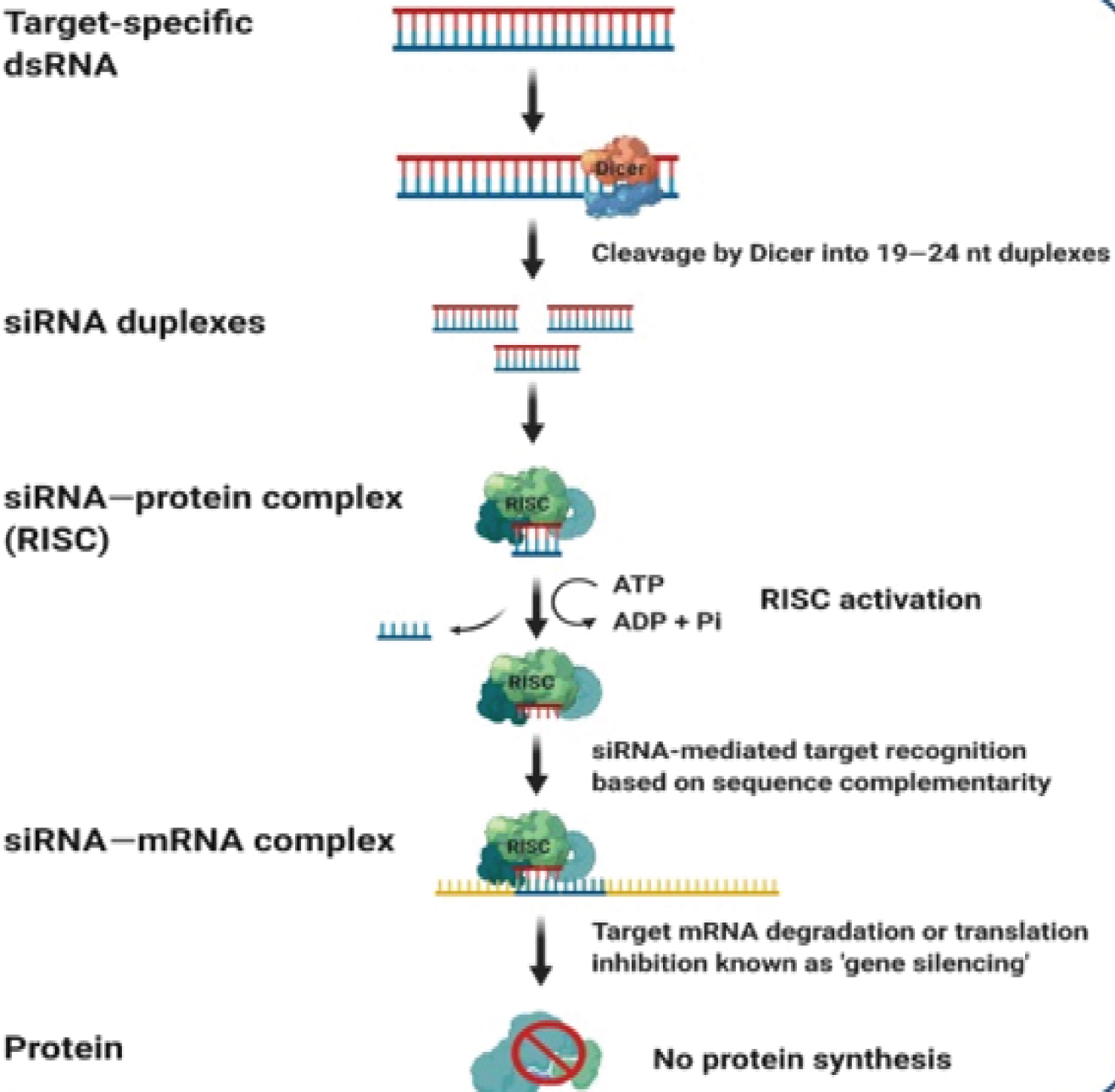 Figure 1: siRNA mediated gene silencing [6].
Figure 1: siRNA mediated gene silencing [6].
siRNA: Small Interfering RNA; RISC: RNA Induced Silencing Complex
View Figure 1
siRNA-based therapeutics use siRNA which is a noncoding double stranded RNA (dsRNA) molecule containing phosphorylated 5' and hydroxylated 3' ends having two unpaired overhanging nucleotides at 3' ends. siRNAs were firstly discovered in plants with their role in post transcriptional gene silencing (PTGS) by a group of scientists at The Sainsbury Laboratory (TSL) in England [7]. An enzyme Dicer (endoribonuclease dicer) is a helicase with RNase motif, encoded by DICER1 gene, forms siRNA by cutting down long dsRNA that enables the formation of RNA induced silencing complex (RISC) [8]. siRNA is incorporated into RISC and induces mRNA-degradation (homology dependent) of cognate mRNA. As siRNA is incorporated into RISC, the passenger strand of siRNA is degraded and removed out of RISC while the remaining strand acts as a guide strand that induces mRNA cleavage (that directs the degradation of targeted mRNA) and is fully complementary to mRNA target. This degradation of mRNA occurs after transcription, preventing translation by silencing the gene expression that results in reducing translation of mRNA into amino acid and inhibits the overproduction of key proteins [9]. In case of MI, such key protein is apolipoprotein (a) which has recently been declared as a highest risk factor for genetically premature MI as well as other CVDs [10]. Reduction in Apo(a), unable the formation of LP(a) due to not binding with low density lipoprotein (LDL) and thus lowering CVDs (Figure 2).
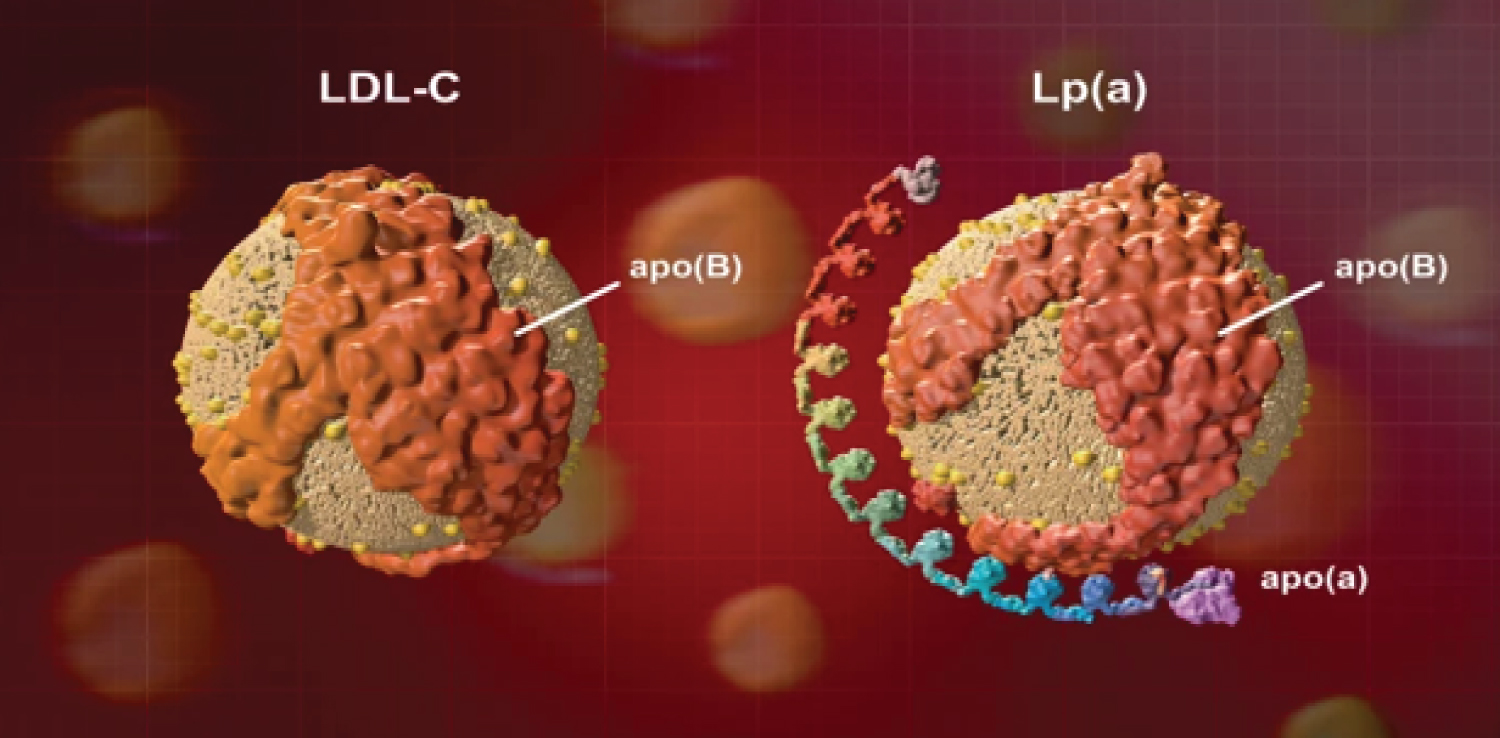 Figure 2: Structure of LP(a) and LDL-C.
Figure 2: Structure of LP(a) and LDL-C.
LDL-C: Low Density Lipoprotein Cholesterol; Apo: Apolipoprotein; LP(a): Lipoprotein(a)
View Figure 2
Few people know about the risk of heart disease from high Lp(a). Diet and exercise will not help because genes control how much LP(a) a person makes. Credit: Craig Keifer/Amgen [11].
LP(a) is a plasma lipoprotein consisting of LDL particle and an additional protein called apolipoprotein (a) [Apo(a)] that is attached to LDL via disulfide bond. LDL particle is also covalently attached to apolipoprotein B [Apo(b)]. LP(a) level in blood plasma is highly heritable via LP(a) gene present at chromosome6q-26-27. Apo(a) surrounds LP(a) molecule and inhibits it to interact with LDL receptors to lower cholesterol level. It causes elevation of inflammatory oxidized phospholipids having high concentration of LP(a) particles resulting in atherogenesis or coronary heart diseases (CHDs) [12]. Polymorphism in LP(a) gene (OMIM152200) codes to produce more Apo(a) level in LP(a). Kringle IV type 2 (KIV-2) polymorphism is one of the most influential LP(a) polymorphisms that results in the multiple formation of Apo(a) isoforms having varied sizes. LP(a) level is inversely correlated with these large number of Apo(a) isoforms (KIV-2 repeats) [13]. Association of high level of LP(a) gene (hyperlipoproteinemia) with increased risk of MI has already been proved using real time PCR assays to genotype for LP(a) KIV-2 repeat polymorphism in LP(a) gene [14]. This association has also been supported by Mendelian randomization analysis [15].
According to a survey, roughly 20% of European population has elevated level of LP(a). National Heart Lung and Blood Institute (NHLBI) has also reported in 2018 that around 1.4 billion people have LP(a) level greater than 50 mg [16]. LP(a) level in blood plasma is mainly dependent on the genetic factors (random distribution among males and females) and the nutritional factors or diet has a very low effect on the increase of LP(a) levels [17]. Enzyme-linked immunoassays (ELISA), especially isoform-insensitive ELISAs have been discovered as reference standard to measure LP(a) levels and fresh serum with ELISA has also reported a very strong relationship of increasing CVDs with elevated LP(a) levels that increased the research interest of correlation between MI and LP(a) levels [18]. Various procedures (excluding RNA-based) for decreasing LP(a) level are being administered clinically, but none of them can eradicate more than 50% of LP(a) of blood plasma. RNA interfering (RNAi) therapies have recently been proved effective to cure acute genetically transmitted MI. This review highlights the potential risk of MI based on genetically acquired elevated LP(a) levels and explains siRNA mediated gene silencing focusing on LP(a) as a target. It also indicates how siRNA therapy [targeting LP(a)] is the most efficient treatment to reduce inherited risk of premature AMI and related CVDs.
The association between elevated LP(a) levels and general CVDs has also enlightened the genetic risk of MI, as elevated LP(a) levels in blood plasma (associated with Apo(a) isoform) observed frequently among the CVD and coronary artery disease (CAD) patients [19]. This association also progressed the research on LP(a) gene that weather LP(a) levels are majorly controlled by the alleles located on hypervariable Apo(a) gene locus or the other factors such as environment and diseases may also have effect on elevating LP(a) levels. Six populations from different areas as well as different ethnic groups have been studied to observe LP(a) levels among MI patients from these different populations. It was observed that LP(a) levels were high in some of the populations having MI patients while the patients from some of the populations shown normal LP(a) levels which demonstrates clearly that LP(a) levels are mainly determined genetically by Apo(a) alleles and other environmental factors have very low effect on LP(a) elevation that results in AMI [19]. Chromosomal regions of LP(a) gene (associated with MI and related coronary diseases) are 6q26-27, 9p21 and 1p13. A study conducted to identify the genetic determinants of LP(a) that are responsible for MI revealed two common variants, rs10455872 and rs3798220 [20]. These variants are found to be strongly associated with elevation of LP(a) lipoprotein that has a direct effect on AMI risk elevation. A comprehensive study conducted to observe correlation LP(a) levels and MI has also revealed that genetically elevated LP(a) levels (two times from normal level) can increase the risk of MI to 22% [21]. Instead of causing stable angina or long-term CVDs, Elevated levels of LP(a) gene usually results in sudden heart stoke or AMI among young and healthy people [22]. Therefore, genetically transmitted elevated LP(a) level is considered as one of the highest risks of sudden heart failure without showing some early symptoms to eliminate risk of premature AMI before it's too late.
Although elevated LP(a) level is variable among different population in all over the world, yet it has become a global concern as it has spread out throughout the whole world. The migration track in Figure 3, clearly demonstrates the worldwide spread of elevated LP(a) levels (> 50 mg/dL). It has also shown that LP(a) gene with different isoforms is more prevalent in Africa as it is also considered to be the origin of elevated LP(a) gene, while East Asia and North America has lowest population of elevated LP(a) levels.
 Figure 3: Migration of LP(a) gene from Africa to rest of world [16].
View Figure 3
Figure 3: Migration of LP(a) gene from Africa to rest of world [16].
View Figure 3
A study conducted by multi-ethnic study of atherosclerosis (MESA) based on the samples (ethnically diverse) of more than 6000 Americans (between 45-84 yr aged) has also found the increased risk of MI with elevated levels of LP(a) [23]. This study has also shown that Black Americans are ay the higher risk of CHDs as well as MI due to higher LP(a) levels. So far, MESA studies have not found this immense risk of genetically transmitted elevated LP(a) levels among the Asian-Americans or Chinese descendants [23]. This shows that the genes carrying higher LP(a) levels have already been transferred from Africa to America because of large amount of immigrants' settlement of Africans who migrated from Africa to America in 16th and 17th century. Although there is not any high level of MI risk [associated with inherited LP(a) levels] found among Asians yet, but the historical data of migration of elevated LP(a) gene indicates the high level of concern among the Asians and Europeans. Recently, a meta-analysis based on European samples (> 50,000) has also revealed that people having LP(a) levels greater than 50 mg/dL are showing new types of CVDs leading to MI and heart stokes as compared to people having LP(a) levels lower than this threshold [24].
In the initial years, after the discovery of LP(a), researcher found that high LP(a) levels are caused by LP(a) sequencing and coding for Apo(a). At that time, no evidence was discovered yet about the genetic evidence that LP(a) is not just a biomarker for high levels, but also a genetic causal factor that is inherited from parents to later generation. Therefore, several therapies (discovered and clinically implemented at that time to lower LP(a) levels) were not based on RNA. These therapies include low dose aspirin therapy, cascade screening, high intensity stain therapy and LP(a) apheresis therapy. Although these therapies can be helpful in lowering LP(a) levels, but are not considered super-efficient in reducing the risk of MI. For example, low dose aspirin therapy cannot reduce the risk of MI among non-carriers and is not suitable for the people having high elevated LP(a) levels (> 50 mg/dL) but no CVD symptoms [25]. Several RNA based treatments are either under clinical trials or are being administered clinically. For example, Inclisiran is GalNAc-modified siRNA-based therapy that targets proprotein convertase kexin type 9 (PCK-9) to lower cholesterol level in plasma and results in lowering the risk of atherosclerosis leading to MI and other related CVDs [26]. It has been studied that, although this strategy of decreasing cholesterol can reduce non-HDL-C (45%), apolipoprotein-B (41%) and overall plasma cholesterol (37%), but the overall reduction in CVDs Including MI is only 24%. Also, this strategy does not mainly target inherited LP(a) gene to reduce its expression of elevated LP(a) levels [26]. ARO-ANG3 is another siRNA-based therapy that targets angiopoietin-like protein 3 (ANGPTL3) and under the clinical trials (NCT03747224) [27]. This ANGPTL3 inhibition therapy reduces LDL levels from 39 to 40% and gives hope to treat hyperlipidemias and CVDs. But with the reduction of LDL levels, this therapy also reduces HDL-C level by 30% [27]. Therefore, this treatment is not ideal for homozygous familial hypercholesterolemia (HoFH) to prevent CVDs. Another RNAi based therapy is APOC-III therapy, that targets APOC-III and regulates Triglyceride (TG) hydrolysis by reducing LDL-C. It results in up to 70% reduction in TG level and can be helpful in prevention of CVDs [28]. But this therapy is also not much effective as it does not reduce the genetic risk of MI by targeting LP(a) and does not cause much reduction in the risk of CVDs. This therapy has also showed some side effects such as vascular adhesion and increased accumulation of inflammatory cells in vascular walls [29].
Although, all these RNA-based therapy have some positive effect in reducing the risk of MI but none of them are effective in reducing inherited risk of MI due to LP(a) elevation. To target genetically elevated LP(a) levels, LP(a) gene need to be silenced to produce less LP(a). Therefore, another siRNA-based therapy has recently been developed to target LP(a) gene and reducing genetic risk of MI. It has been observed that PCKS9 inhibitor evolocumab can also reduce LP(a) plasma levels by 25%, but this is not enough to eradicate the risk of AMI [30]. Olpasiran (AMG-890) is GalNAc-modified siRNA-based therapy that targets LP(a). It is blunt ended 19-mer mixed 2'-O-Me targeting mRNA of LP(a). It is modified with 2'-fluoro and 2'-methoxy substitutions and phosphonothioate inter-nucleotide linkages. Phase II trials (NCT03626662, NCT04270760) are being tested and the initial results have shown that it can reduce LP(a) levels by 80-94% at day 113 with no side effects [31]. Reduction of LP(a) levels to this extent has not been observed by any of the therapy other than siRNA-silencing LP(a) therapy. This therapy is useful to reduce genetically transmitted LP(a) levels which is mainly based on genetically transmitted information and not on diet and exercise. Therefore, it can not only be helpful for the patients having risk of AMI but can also protect the upcoming generations from premature MI.
Experimental plan of siRNA-based gene silencing consists of three major processes. First stage is to design the experiment. It includes selecting the appropriate biological model by knowing the expression of targeted gene as well as its nucleotide sequence. Then the siRNAs are generated specifically for the target gene according to complementary sequence, G/C content, length and positions of nucleotides and gene specificity. A negative control should be established to check the validity of knockdown by siRNA to target gene. This can be done by creating non-specific siRNA sequences with targeted gene [9].
Next stage is to deliver siRNA into the targeted cells. It is mainly achieved by transfection processes such as lipofection in which lipid mixture mixed with micelles (to get physiological pH) form complex with nucleic acids of siRNA. This mixture is applied on the targeted cells. Liposomes or micelles attach and fuse with cell membrane that results in penetration of siRNA into the cell to interact mRNA present in the cell's cytoplasm [9]. Optimization of siRNA delivery into the cell can be done to achieve efficiency, by various ways such as optimizing siRNA/liposome ratio, amount and health of targeted cells, incubation time and temperature of transfection. Efficiency and toxicity of transfection can be identified by fluorescently labeled siRNA (siFlu) and comparing the number of cells survived and transfected with the control group respectively. If the transfection efficiency is more than 70%, it is suitable for knockdown by siRNA which includes, mixing the cell culture, siRNA (combined with appropriate micelles) and metafectene. Incubating the mixture at room temperature into wells with 5% CO2 for 24 hours can allow the knockdown by siRNA to degrade the mRNA of targeted cells [9].
The third stage is to evaluate the knockdown efficiency that include the isolation of total RNA from transfected cells, treatment of RNA with DNase I to remove DNA contamination, quality control of DNase I efficiency, cDNA synthesis by reverse transcription, quality control of cDNA synthesis efficiency and evaluating the gene expression by real time PCR assays [9].
siRNA based Olpasiran silences the expression of LP(a) gene by mRNA degradation to reduce the Ap(a) protein production and LP(a) particles in hepatocyte. It is targeted to liver via GalNAc moiety and then gets attached to asialoglycoprotein receptors (ASGPR) on the surface of hepatic cells (Figure 4) [32]. As Olpasiran gets inside the hepatic cells (hepatocytes), one of its strands (antisense strand) enters in RISC while the other (sense strand) strand is degenerated. RISC, loaded with antisense strand gets attached to mRNA of Apo(a). The binding is complimentary due to its antisense strand of Olpasiran. mRNA gets degraded due to argonaut proteins associated with RISC complex [33]. This mRNA degradation is carried out further as RISC complex disassociate itself from degraded mRNA and binds with another mRNA to repeat this process. In this way it silences the LP(a) expression to build more Apo(a) thus reducing LP(a) levels and hence AMI.
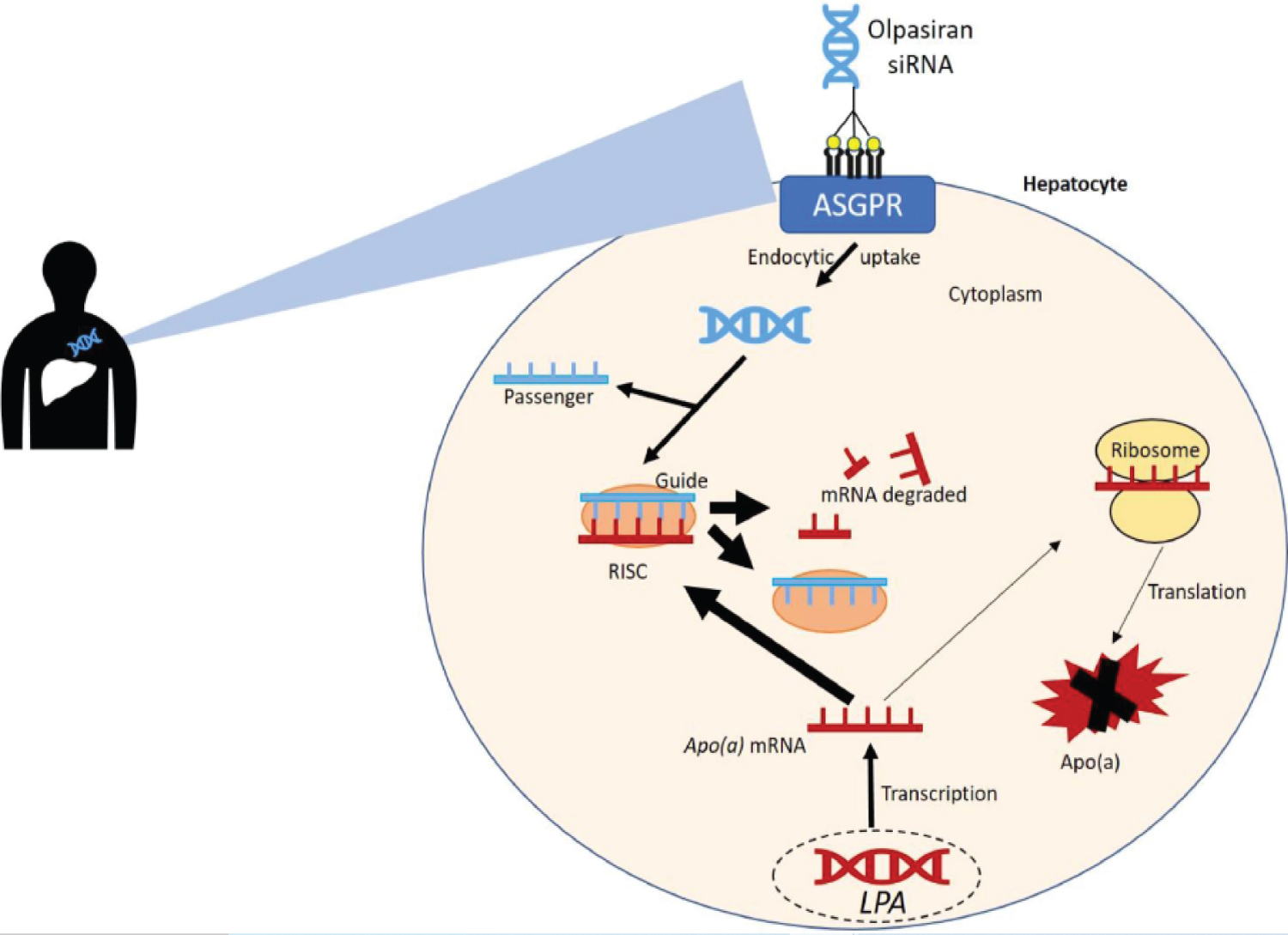 Figure 4: Mechanism of Olpasiran (siRNA-based therapy) to reduce LP(a) levels [32].
View Figure 4
Figure 4: Mechanism of Olpasiran (siRNA-based therapy) to reduce LP(a) levels [32].
View Figure 4
Pelacarsen is another therapy by Novartis that targets LP(a) to reduce Apo(a). It is 2'-methoxyethyl chimeric antisense oligonucleotide (ASO) ARO-APO(a)-LRx that is thought to reduce LP(a) levels by 80% [34]. Like Olpasiran, Pelacarsen is also attached to GalNAc, but it is triantenary (GalNAc3) especially developed by Novartis and Akcea therapeutics. It also prevents the translation of mRNA and inhibits the production of Apo(a) but unlike siRNA-therapies that contain both antisense and sense strands, ASO-drugs are single stranded RNA or DNA molecules. Phase 2 trials of Pelacarsen have shown a few side effects while the Phase 3 trials are under evaluation that started in 2020 and expected to end in 2025 which is still three years away. London-based Silence therapeutics has reported another therapy named as SLN-360 is also siRNA-based therapy that targets LP(a) like Olpasiran and according to its Phase 1 trial, SLN-369 is believed to reduce LP(a) levels up to 90% [35]. Mechanism of SLN-360 to reduce LP(a) levels is also similar to Olpasiran's mechanism.
As shown in the Table 1, LP(a) level is mainly reduced by LP(a) targeted therapies. But the strategy of targeting LP(a) is not enough to reduce much level of LP(a). For example, Lipid apheresis therapy is also LP(a) targeted therapy, but the reduction of LP(a) level is only 35%. siRNA-based LP(a) targeted therapies are the most effective therapies to reduce high levels of LP(a) (up to 97%). These include Olpasiran and SLN 360 and both are based on siRNA-based LP(a) targeted therapy. Although ASO-based LP(a) targeted therapy (Pelacarsen) is also effective in lowering LP(a) levels which is up to 80%, but still this level is lower than siRNA-based therapies. Moreover, the results of phase 2 trial of Pelacarsen are still three years away, while the latest phase 2 results of siRNA-based therapy (Olpasiran) have been recently shown by Amgen that seems to be most efficient reduction in LP(a) levels among all the rest of known therapies based on mechanisms other than LP(a) targeting.
Table 1: Various drugs/therapies and their effect on LP(a) reduction [11,36]. View Table 1
CVDs are the most common diseases of today's world and MI is one of leading causes of death among CVD patients. Although there are several therapies and drugs available to reduce the risk of MI, but the genetic risk of MI (which has recently been estimated as half of the total MI cases) is still not perfectly treatable as it is due to genetically elevated LP(a) levels and diet & physical activity has almost no effect on it. Elevated LP(a) levels has already been spread throughout the world and is estimated to be transferred rapidly in upcoming generations. Several RNA based therapies has been clinically tested to lower down LP(a) levels and some of them are under trials. siRNA-based therapy (that is based on lowering LP(a) by degrading mRNA to inhibits Apo(a) production) has recently shown the most effective results [97% reduction in LP(a) levels] among all the rest of known therapies and can be the most ideal choice to reduce the inherited risk of MI. Although siRNA-based LP(a) silencing therapy (Olpasiran) has not been approved by FDA yet, but its recent Phase II results have proved that siRNA-based gene therapies can not only be the future of premature and inherited AMI treatments but also protect the future generations. The golden era of siRNA therapeutics is on the horizon but still it has been at least three years before any of the siRNA-based LP(a) silencing therapies can reach the market [11]. Therefore, until then, a huge educational effort is required to motivate people for normalizing the screening of familial markedly elevated LP(a) levels. Medical authorities and governmental bodies also need to accelerate the efforts for getting siRNA therapies approved, available and affordable for public as soon as possible. Although, European guidelines recommends LP(a) levels of 50 mg/dL or higher than this threshold is a potential target for screening and treatment, but this suggestion is still absent in United States' guidelines.
The author has received no financial or academic support for the research, authorship, and publication of this article.
None to declare.
This author takes responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.