Background and objective: The details of thrombolytic activity in occlusive disease associated with atherosclerotic lesions are not clear. This study investigated the novel effects of activated coagulation factor X (F-Xa)-directed oral anticoagulant (DOAC) edoxaban on in vivo thrombolytic activity and atherosclerosis progression.
Methods: Mice were fed an experimental diet (edoxaban 15-50 mg/kg) for 18 weeks. The degree of progression of atherosclerosis was assessed as the area of atherosclerotic vessels as a percentage of the total area of vessels. In vivo thrombolytic activity was determined by measuring the change in volume of thrombus formed by He-Ne laser. Blood vessels and livers were excised and subjected to immunostaining and real time PCR analysis of relevant genes.
Results: Edoxaban treatment did not affect the progression of atherosclerosis and did not affect the expression levels of relevant genes (IL-6, TNF-α, MCP-1, MMP-9, EGR-1 and VEGF-α genes). In contrast, in vivo thrombolytic activity was significantly enhanced in the edoxaban 50 mg group compared to the placebo group. The expression of t-PA mRNA in vessels was significantly increased in the edoxaban group and the expression of TAFI by immunostaining were significantly decreased.
Conclusions: Edoxaban did not affect the development of atherosclerosis, but promoted thrombolysis in vivo accompanied by increased t-PA mRNA levels and decreased local TAFI expression. This was considered evidence suggesting that a novel action of F-Xa-directed DOACs is to enhance spontaneous fibrinolytic activity.
Edoxaban, Factor-Xa, Thrombolysis, Atherosclerosis, Thrombin
Atherosclerosis is the etiology of ischemic heart disease and cerebrovascular disease, and is one of the leading causes of death in developed countries [1]. The progression of atherosclerosis is accompanied by tissue lesions, forming plaques with lipid accumulation beneath the endothelial stroma. These plaques are characterized by angiogenesis and are prone to rupture [2,3]. Ischemic disease due to thrombotic occlusion caused by plaque rupture can lead to serious consequences. It is important to recanalize the occlusion by thrombolytic therapy as soon as possible, and its treatment determines the prognosis. Since thrombin, which plays a central role in the coagulation system, and F-Xa, which is directly involved in its production, are involved in the formation of occlusive thrombi, oral direct thrombin inhibitors and oral direct F-Xa inhibitors (DOACs) have been developed as anticoagulation therapy to prevent thrombus formation. F-Xa is directly involved in thrombin production and is therefore a target for anticoagulation therapy, leading to the development of the F-Xa-directed DOAC edoxaban as an alternative to warfarin [4]. Currently, edoxaban [5] as well as rivaroxaban [6], and apixaban [7] are available for clinical use.
We previously reported that long-term treatment with the thrombin-directed oral anticoagulant Dabigatran in Apoe-/- and Ldlr-/- double knockout mice not only inhibited the development of atherosclerosis but also promoted thrombolysis in an in vivo model [8]. This result suggested the possibility of DOACs as a new additional action. Like thrombin, F-Xa has many physiological and pathological effects such as immunity, inflammation, and wound healing [9,10] through its involvement in the PAR-1 and PAR-2 signaling pathways as well as in the coagulation reaction. Since edoxaban directly inhibits F-Xa, we hypothesized that edoxaban, an F-Xa-directed DOAC, may have an effect on spontaneous thrombolytic activity and on atherosclerosis development, in addition to F-Xa inhibition as well as thrombin-directed DOAC.
The aim of this study was to test the potential new action of the F-Xa-directed DOAC, edoxaban, by examining how it affects spontaneous thrombolytic activity in an in vivo model and its effect on the progression of atherosclerosis in Apoe-/- and Ldlr-/- double knockout mice.
C57BL/6J × 129Sv double-knockout male mice lacking both apolipoprotein E and LDL receptor (B6.129-Apoetm1Unc Ldlrtm1Her/J miceApoe; hereafter, DK mice) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and C57BL/6J male mice were purchased from SLC Japan (Hamamatsu, Japan). Mice were kept in chip cages (Softchip; SLC Japan (Hamamatsu, Japan) in the Kobe Gakuin University animal room (light and dark 12 hours each, room temperature 22.5 ± 0.5 °C, and humidity 55 ± 5%). The animals were given free access to tap water and standard solid feed (CMF; Oriental Yeast Co., Ltd., Tokyo, Japan).
The handling of experimental animals was in accordance with the Basic Policy on Animal Experiments in the Field of Physiology established by the Physiological Society of Japan and the Kobe Gakuin University Policy on Bioethics in Research and Education.
The oral direct F-Xa inhibitor edoxaban (edoxaban tosilate hydrate) was supplied by Daiichi Sankyo Company Limited (Tokyo, Japan).
Experimental diets were prepared from standard solid feed (CMF; Oriental Yeast Co.) supplemented with 0, 15, 30, or 50 mg/kg edoxaban. Agar (final concentration 2%; Fujifilm Wako Pure Chemical Co., Ltd., Osaka, Japan) was added to the feed mixtures to make them solid, and they were kept frozen at -20 °C until used in experiments.
DK and C57BL/6J mice were fed unmodified CMF until 4 weeks of age, then assigned to groups which were each fed the corresponding experimental diet for 18 weeks. At 22 weeks of age, progression of arterial stiffness and spontaneous thrombolytic activity were measured, blood was sampled, and the ascending aorta, brachiocephalic artery, left subclavian artery, aortic valve, and liver were removed for various assays.
Blood was collected from the abdominal aorta using a 24G needle in a 1.5 mL microtube with 1/10 volume of 3.14% sodium citrate solution placed in the syringe in advance. Immediately after collection, the blood was centrifuged (3000 rpm, 10 min), and the plasma was frozen at -30 °C until measurement. At the time of measurement, plasma was thawed in a thermostatic bath at 37 °C and measured with a Biophen Edoxaban Calibrator (Hyphen Biomed, Oise, France).
The tail-nick method was used to assess bleeding time. A disposable blood lancet (Feather, Tokyo, Japan) was used to make a nick approximately 1.5 cm from the base of the tail of anesthetized mice. The hemorrhaging blood was blotted with a piece of filter paper, and the time until the blood stopped adhering to the filter paper was measured.
Plasminogen activator inhibitor 1 (PAI-1) was measured using a Total Mouse PAI-1 ELISA Kit (Innovative Research, Inc., Novi, MI, USA), tissue-type plasminogen activator (t-PA) was measured using a Mouse tPA ELISA Kit (Elabscience, Houston, TX, USA), and thrombin activatable fibrinolysis inhibitor (TAFI) was measured by ELISA with a Total TAFI ELISA Kit (Innovative Research, MI, USA). The same frozen plasma used for the edoxaban measurement in blood was used as the sample.
Staining of atherosclerotic areas: The entire aorta method [11] was used to measure development of atherosclerosis. Anesthetized mice were subjected to transcardial perfusion with PBS and 10% neutral buffered formalin, and the entire aorta was removed. The vessel lumen was inverted by means of a longitudinal incision, and the vessels were spread open over a black rubber sheet and held in place using 0.15 mm diameter stainless steel pins. The pinned vessels were stored in 10% neutral buffered formalin fixative until staining. For staining, specimens were washed with distilled water for 30 seconds, immersed in 60% isopropyl alcohol for 1 minute, and then immersed in Oil Red O (Sigma-Aldrich Chemie GmbH, Schnelldorf, Germany) pre-warmed for 15 minutes at 37 °C. Following staining, the specimens were immersed in 60% isopropyl alcohol for 2 minutes and washed with distilled water for 2 to 3 minutes. Analysis of atherosclerotic area: The entire surface area of the extracted blood vessel (W) and the area stained red with Oil Red O (R), which corresponds to the atherosclerotic area, were determined by using image analysis software (Image-Pro Plus; Media Cybernetics, Inc., Rockville, MD, USA). From these two values, we calculated the percentage of the entire blood vessel that was affected by atherosclerosis [(R/W) × 100], and this was defined as the atherosclerotic area.
A polyethylene tube (PE-10; Becton Dickinson, Franklin Lakes, NJ, USA) was placed in the left femoral artery of mice for intravenous infusion of Evans blue. Blood vessels running over the cremaster muscle of the mice were then exposed and secured with clips. Tyrode’s solution at 37 °C was dripped onto the vessels from time to time to prevent desiccation. Following this operation, the mice were fixed on a microscope stage (Model BX; Olympus Co., Ltd., Tokyo, Japan) in a chamber heated to 37 °C. A 30-μm-diameter vessel running over the cremaster muscle was selected, and a He-Ne laser (DPS-5002; NEOARK, Tokyo, Japan) with an output power of 20 mW was focused to 5 μm below the objective lens; the vessel was irradiated for 5 seconds, followed by a 25-second interruption. This was repeated until a stable mural thrombus had formed that stenosed 80% of the target vessel lumen.
After the stable mural thrombus that stenosed 80% of the vessel lumen had formed, the thrombus was video-recorded for 60 minutes using a DVD recorder (DVR-710H; Pioneer, Tokyo, Japan). The recorded thrombus images at several time points (0, 10, 20, 30, 40, 50, and 60 minutes) were captured on a PC, and the thrombus size was estimated by using Image-Pro Plus image analysis software. The thrombus volume at time of thrombus formation (minute 0) was set as 100%, and the thrombus volume at each subsequent time point (10, 20, 30, 40, 50, and 60 minutes) was calculated as a percentage relative to the initial volume.
C57BL/6J mice and DK mice were opened at the chest and abdomen, a winged needle was inserted into the left ventricle, and the vasculature was flushed with PBS (pH 7.4) for approximately 3 minutes to wash out the blood. The carotid artery and right and left subclavian arteries were then severed and the thoracic aorta was cut from the heart. Adipose and connective tissues attached to the vessels were carefully removed. The vessel to be sampled (ascending aorta) was cut into sections, immersed in OCT compound, and flash-frozen in dry ice and acetone mixture. Frozen specimens were sectioned to 10 μm thicknesses by cryostat, placed on coated glass slides, wrapped in aluminum foil, and stored at -30 °C until staining.
After bringing the glass slides to room temperature, they were fixed with acetone, washed with PBS, and antigen-activated with 10-fold-diluted Target Retrieval Solution (Agilent: CA, USA) at 60 °C for 5 hours. After washing with PBS, a drop of blocking reagent was applied to cover the entire section to inhibit endogenous peroxidase and allowed to react for 10 minutes at room temperature. After washing with distilled water and PBS, the sections were incubated with the following antibodies (each diluted 100-fold) at 4 °C overnight: anti-PAI-1 rabbit polyclonal antibody (Bioss, MA, USA); anti- matrix metalloproteinase 9 rabbit (MMP) polyclonal antibody, gelatinase B, 92 kD type IV collagenase (BioVision, CA, USA);, anti-Thrombin activatable fibrinolysis inhibitor (anti-TAFI) antibody (1:100; TAFI Rabbit PAb, GeneTex, Irvine, CA, USA) and anti-t-PA rabbit polyclonal antibody (Technoclone, Vienna, Austria). After washing with PBS, anti-rabbit Ig pig polyclonal antibody (Agilent, CA, USA) was added dropwise as a secondary antibody and allowed to react for 40 minutes. After washing with PBS, a chromogenic substrate solution (Liquid DAB+ Substrate Chromogen System (Agilent)) was added dropwise and allowed to react for 5 minutes at room temperature. After washing with distilled water, nuclear staining was performed with Mayer's hematoxylin (Muto Chemical Co., Ltd., Tokyo, Japan) for 3 minutes. After rinsing, the samples were dehydrated, permeabilized, and sealed with marinol.
To evaluate the relative density of stained areas, staining intensity was quantified by using Image Pro Plus digital image analysis software according to the manufacturer’s immunochemical data analysis protocols. Staining intensities were inversed for sections of fixed areas containing atherosclerotic plaques, and were averaged for each mouse.
RNA extraction: Mice were anesthetized and abdominally opened, and blood was collected from the common iliac artery bifurcation of the abdominal aorta to obtain citrated plasma. A 21G winged needle was then inserted into the left ventricle, and 30 mL of PBS was circulated to wash out the blood.
The ascending aorta, descending aorta, liver, and kidneys were then collected, weighed, flash-frozen in liquid nitrogen, and stored frozen at -80 °C.
Total RNA was extracted from stored samples using a RNeasy Fibrous Tissue Kit (Qiagen, Venlo, Netherlands). The RNA purity and concentration were verified using a NanoDrop One microvolume ultraviolet-visible spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) with absorbance measured at 260 and 280 nm.
Quantification of mRNA Expression: cDNA was synthesized from RNA samples using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). Gene-specific primers and TaqMan probes for MMP-9, TAFI (Cpb2), t-PA (Plat), PAI-1 (Serpine1), IL-6, TNF-α, MCP-1 (Ccl2), Egr1, VEGF-α, and β-actin (Actb) were purchased from Thermo Fisher Scientific, and expression of mRNAs was evaluated using quantitative RT-PCR analysis in a Roche Light Cycler 96 (Roche Diagnostics K.K., Tokyo, Japan). PCR amplifications were performed under the following conditions: one cycle at 50 °C for 2 minutes and 95 °C for 2 seconds, followed by 45 cycles of 95 °C for 3 seconds and 60 °C for 30 seconds. Expression of the mRNA in question was normalized to that of the corresponding β-actin mRNA. Differential gene expression was compared between drug-treated and non-treated groups using the comparative Ct method (ΔCt method) in Roche Light Cycler 96 SW 1.1 analysis software (Roche Diagnostics).
Statistical analysis was performed using the paired t-test between two paired groups, Student's t-test for equal variance between two paired groups, and the Mann-Whitney U test for unequal variance between two paired groups. Results are expressed as mean ± SEM, with P < 0.05 indicating a significant difference.
Body weights were 41.9 ± 0.7 g in the placebo group (n = 14), 36.8 ± 0.6 g in the edoxaban 15 mg group (DK mice fed 15 mg edoxaban per kg of feed) (n = 19), 36.9 ± 0.8 g in the edoxaban 30 mg group (n = 16), and 36.5 ± 1.0 g in the edoxaban 50 mg group (n = 16). At 22 weeks of age, all edoxaban groups showed significant weight loss compared to the placebo group (P < 0.01).
Bleeding time was 231.4 ± 10.2 seconds for the placebo group (n = 14), 313.3 ± 21.5 seconds for the edoxaban 15 mg group (n = 19), 352.5 ± 33.0 seconds for the edoxaban 30 mg group (n = 16), and 320.6 ± 17.3 seconds for the edoxaban 50 mg group (n = 16). The edoxaban group showed a prolonged bleeding time compared to placebo (P < 0.05-0.01). No bleeding symptoms were observed in any group.
The concentration of edoxaban in the blood was 917.8 ± 26.3 ng/mL in the edoxaban 15 mg group (n = 7), 961.4 ± 17.3 ng/mL in the edoxaban 30 mg group (n = 11), and 1009.1 ± 18.3 ng/mL in the edoxaban 50 mg group (n = 8). There was a significant difference between the edoxaban 15 mg group and the edoxaban 50 mg group (P < 0.01).
Platelet counts were 98.6 ± 3.1 × 104/μL in the placebo group (n = 7), 103.1 ± 5.3 × 104/μL in the edoxaban 15 mg group (n = 5), 95.0 ± 4.8 × 104/μL in the edoxaban 30 mg group (n = 6), and 99.9 ± 10.5 × 104/μL in the edoxaban 50 mg group (n = 6). No group differed significantly from the placebo group.
Representative arteriograms and image analysis results for each DK group at 22 weeks of age are shown below (Figure 1). The degree of progression of atherosclerosis was 35.1 ± 8.0% in the placebo group (n = 6), 39.7 ± 8.1% in the edoxaban 15 mg group (n = 6), 37.4 ± 13.4% in the edoxaban 30 mg group (n = 6), and 35.2 ± 5.7% in the edoxaban 50 mg group (n = 6). No group differed significantly from the placebo group.
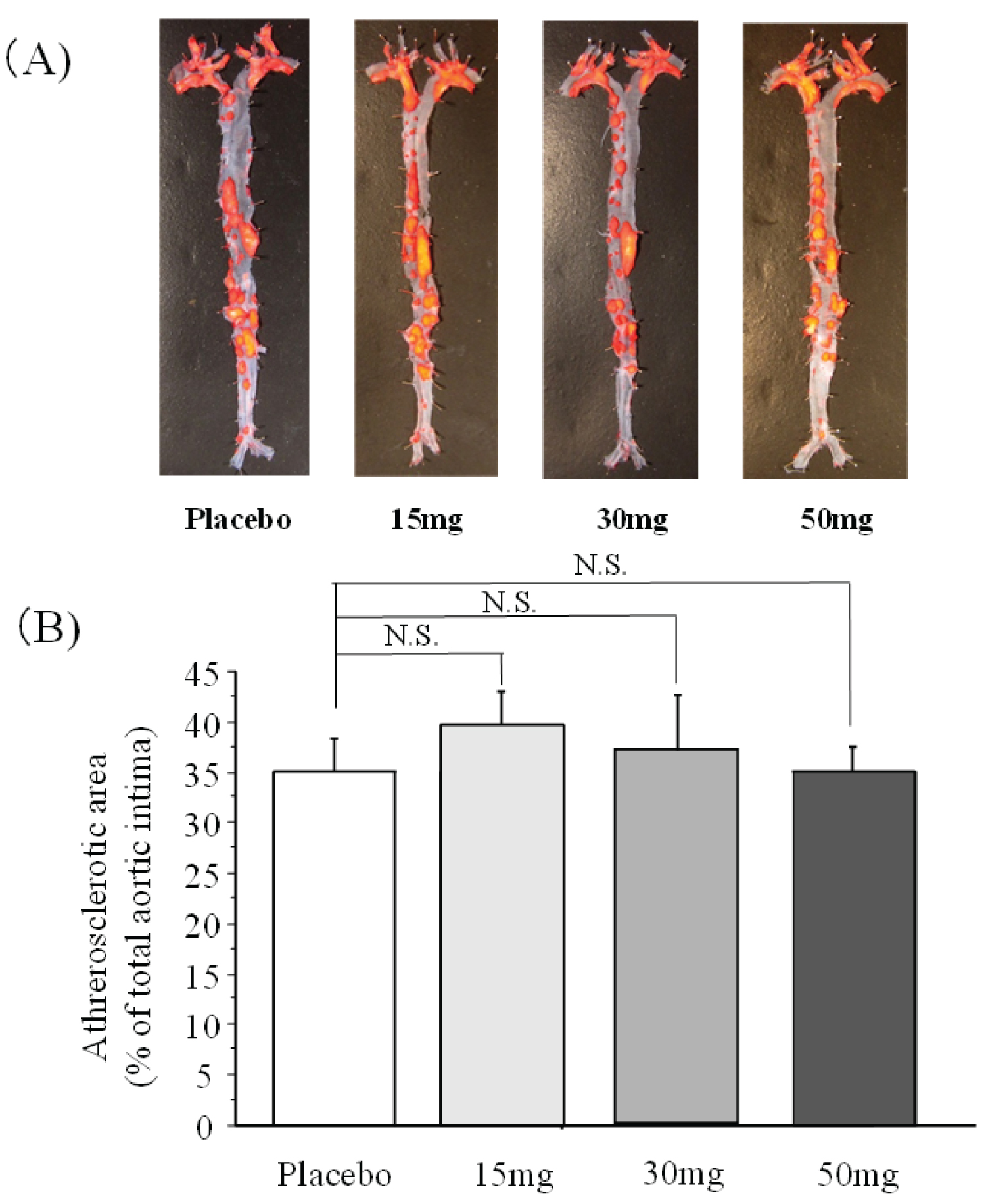 Figure 1: Comparison of atherosclerotic foci (A) Vascular Oil Red O staining; (B) Image analysis results.
n = 6 per group. The label on each column indicates the gloop based on edoxaban concentration (mg) per kg of experimental diet.
View Figure 1
Figure 1: Comparison of atherosclerotic foci (A) Vascular Oil Red O staining; (B) Image analysis results.
n = 6 per group. The label on each column indicates the gloop based on edoxaban concentration (mg) per kg of experimental diet.
View Figure 1
The mRNA expression levels of IL-6, TNF-α, and MCP-1 as inflammation markers related to the development of atherosclerosis, and MMP-9, EGR-1, and VEGF-α as markers related to angiogenesis were measured in the placebo and edoxaban 50 mg groups by real-time PCR. No significant difference in any mRNA expression level was observed between the two groups.
The thrombus volume at 60 minutes as a percentage of the volume of the stabilized thrombus at time of formation was 65.3 ± 5.2% in the placebo group (n = 8), 52.0 ± 6.8% in the edoxaban 15 mg group (n = 7), 63.0 ± 6.5% in the edoxaban 30 mg group (n = 7), and 47.4 ± 5.7% in the edoxaban 50 mg group (n = 8). The thrombus volume ratio at 60 minutes was significantly lower in the edoxaban 50 mg group than in the placebo group (Figure 2).
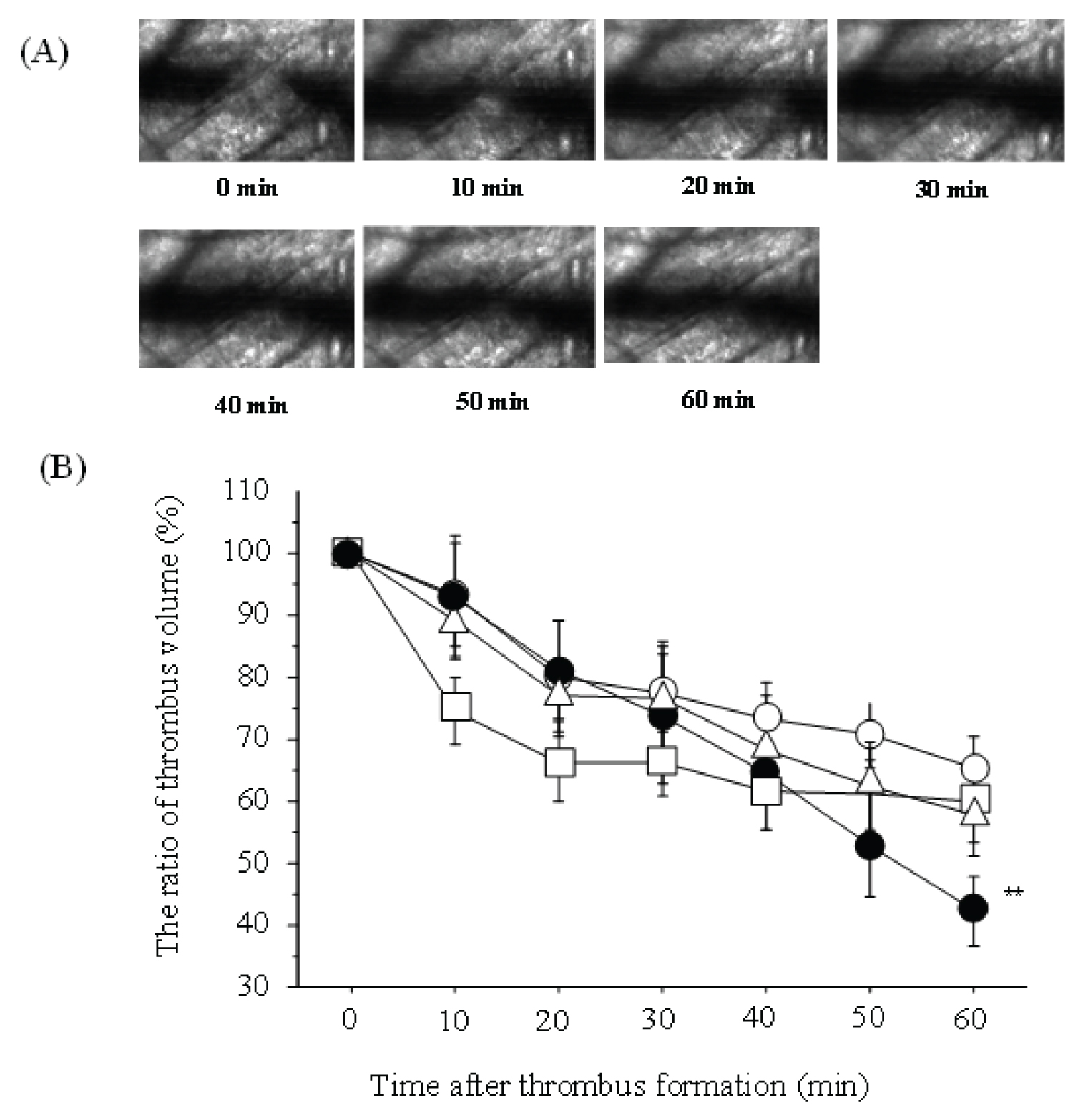 Figure 2: The changes of Spontaneous thrombolysis by edoxaban (A) Typical changes in thrombus volume by edoxaban over time; (B) Thrombus volume as a percentage of initial volume in each group over time.
Figure 2: The changes of Spontaneous thrombolysis by edoxaban (A) Typical changes in thrombus volume by edoxaban over time; (B) Thrombus volume as a percentage of initial volume in each group over time.
○: placebo group, △: edoxaban 15 mg group, □: edoxaban 30 mg group,
●: edoxaban 50 mg group, n = 7-8/group **: P < 0.01 vs. placebo group
View Figure 2
Levels of t-PA in the blood were 727.5 ± 65.9 pg/mL in the placebo group (n = 14), 718.2 ± 57.4 pg/mL in the edoxaban 15 mg group (n = 14), 717.1 ± 50.8 pg/mL in the edoxaban 30 mg group (n = 17), and 706.1 ± 64.7 pg/mL in the edoxaban 50 mg group (n = 14). There was no significant difference between any of the groups.
Total PAI-1 levels in blood were 5.2 ± 0.8 ng/mL in the placebo group (n = 14), 6.2 ± 1.5 ng/mL in the edoxaban 15 mg group (n = 17), 7.7 ± 1.5 ng/mL in the edoxaban 30 mg group (n = 14), and 7.1 ± 1.4 ng/mL in the edoxaban 50 mg group (n = 14). There was no significant difference between any of the groups.
Levels of TAFI in the blood were 104.5 ± 12.4% in the edoxaban 15 mg group (n = 17), 100.0 ± 9.0% in the edoxaban 30 mg group (n = 14), and 86.5 ± 6.7% in the edoxaban 50 mg group (n = 14), when the mean value of the placebo group (n = 14) was 100%. There was no significant difference between any of the groups.
In contrast, although immunohistochemical staining showed no difference in the expression of t-PA and PAI-1 between the placebo group and the edoxaban 50 mg group, the expression of TAFI in the edoxaban 50 mg group was decreased compared to the placebo group. Furthermore, there was no difference between the two groups with respect to PAI-1 mRNA expression in blood vessels or TAFI mRNA expression in the liver. However, t-PA mRNA expression in blood vessels and liver in the edoxaban 50 mg group was significantly higher than in the placebo group (P < 0.01, Figure 3).
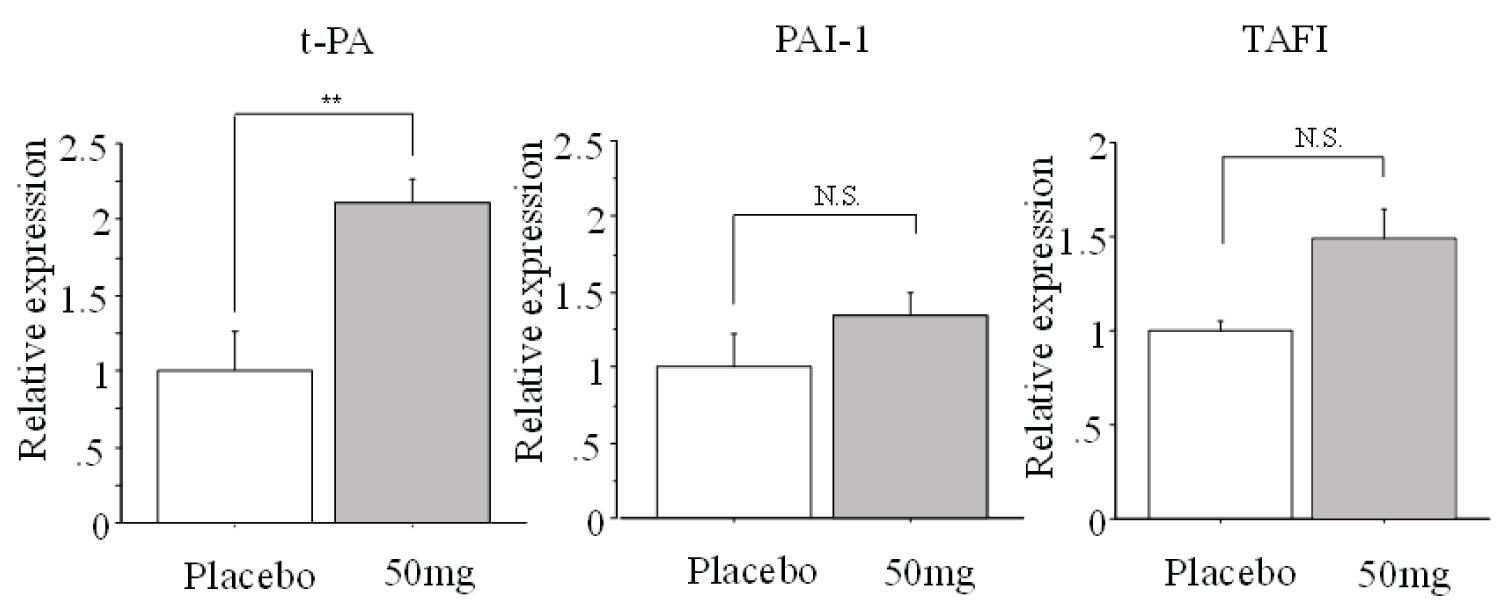 Figure 3: Comparison of mRNA expression of activator t-PA and inhibitors PAI-1 and TAFI in the placebo and edoxaban 50 mg groups.
Vascular samples (t-PA, PAI-1), liver samples (TAFI), n = 6 per group, **: P < 0.01 vs. placebo group.
View Figure 3
Figure 3: Comparison of mRNA expression of activator t-PA and inhibitors PAI-1 and TAFI in the placebo and edoxaban 50 mg groups.
Vascular samples (t-PA, PAI-1), liver samples (TAFI), n = 6 per group, **: P < 0.01 vs. placebo group.
View Figure 3
TAFI staining of the ascending aorta showed that in all groups, TAFI was expressed in and near atherosclerotic plaques (Figure 4). TAFI expression was markedly lower in the edoxaban group than in the placebo groups. There was no difference in expression between the edoxaban groups.
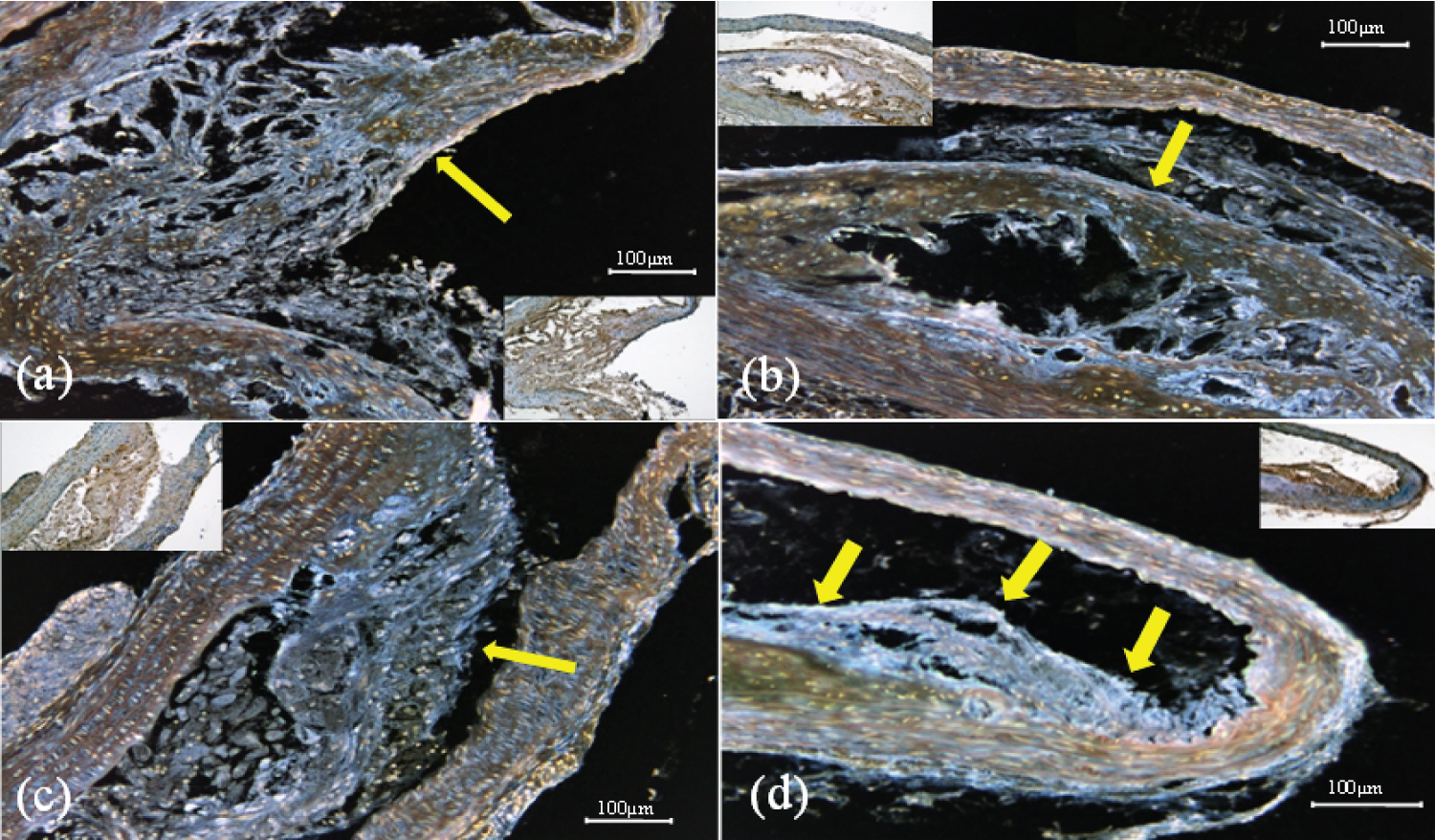 Figure 4: Representative immunostaining images of TAFI expression in each group (a) edoxaban 15 mg group; (b) edoxaban 30 mg group; (c) edoxaban 50 mg group; (d) placebo group Ascending aorta (× 200). Arrows indicate positive areas.
View Figure 4
Figure 4: Representative immunostaining images of TAFI expression in each group (a) edoxaban 15 mg group; (b) edoxaban 30 mg group; (c) edoxaban 50 mg group; (d) placebo group Ascending aorta (× 200). Arrows indicate positive areas.
View Figure 4
Since thrombus formation is a cause of various occlusive diseases, the transition of fibrinolytic kinetics is very important. However, the fibrinolytic kinetics associated with the development of atherosclerosis and other thrombotic pathologies are not clear [12].
We previously reported that under conditions similar to those in the present study, the thrombin-directed oral inhibitor dabigatran markedly inhibited the development of atherosclerosis in Apoe-/- and Ldlr-/- double knockout mice and enhanced spontaneous thrombolytic activity accompanied by decrease immunological expression of PAI-1 and TAFI [8], and reported a possible new clinical action of thrombin-directed DOACs.
In the present study, we investigated how edoxaban, an oral, direct inhibitor of F-Xa, which is directly involved in thrombin generation and has diverse effects, affects the development of atherosclerosis and spontaneous fibrinolytic activity.
Edoxaban, the oral direct inhibitor of F-Xa used in this study, inhibits F-Xa, the starting point of the blood coagulation cascade. Edoxaban competitively inhibits the protease activity of F-Xa with a Ki value of 0.561 nM [4]. It has weak inhibitory activity against serine proteases other than F-Xa, indicating that it is a highly selective inhibitor of F-Xa [13]. Edoxaban is a potent inhibitor of F-Xa, which is the starting point of the coagulation cascade.
Like warfarin and enoxaparin, it has been shown to inhibit venous thrombosis [14] and the risk associated with bleeding is lower with edoxaban than with warfarin or enoxaparin [15] with little effect on bleeding time in a rat tail bleeding time model. Warfarin requires regular monitoring and dose adjustment to maintain the INR (International Normalized Ratio) within the therapeutic range.
In addition, the dose used varies nearly 10-fold from patient to patient, there are many drug interactions, and vitamin K-containing foods must be restricted [16].
Unlike warfarin, edoxaban has a rapid onset of action, does not require monitoring over a relatively wide therapeutic range, is convenient for patients, and is said to be more economical than existing anticoagulants [16]. These conditions requiring DOACs administration are thrombophilic states, and if DOACs were able to stimulate thrombolytic activity, this would be clinically beneficial.
At the doses used in this study, there was a significant decrease in body weight between the edoxaban and placebo groups, but there was no significant difference in platelet counts in the 50 mg edoxaban group, and bleeding time was approximately 1.4 times longer than in the placebo group. However, no cases of intra-abdominal bleeding or hematuria occurred during the study period, suggesting that the risk of bleeding with edoxaban is lower than with warfarin or enoxaparin [17].
Blood edoxaban levels were dose-dependent, although there were no significant differences between the 15 mg and 30 mg Edoxaban groups.
There were no significant differences in the effects of edoxaban treatment on the progression of atherosclerosis between the placebo and edoxaban groups. There were no significant differences in mRNA expression of IL-6, TNF-α, MCP-1 gene (inflammation marker), MMP-9, EGR-1, and VEGF-α gene (mediator of angiogenesis) associated with the progression of atherosclerosis between the placebo and edoxaban 50 mg groups, suggesting that edoxaban had no effect on atherosclerosis-related genes.
These results supported the report that reversaloxaban, the same oral direct F-Xa inhibitor as edoxaban, did not inhibit plaque progression in atherosclerosis [18].
The lack of inhibition of atherosclerosis progression by F-Xa inhibition differs from the previously reported results with the thrombin inhibitor dabigatran [8].
On the other hand, we investigated the effect of edoxaban on spontaneous thrombolysis in an in vivo thrombolysis model. In the change in thrombus volume at 60 minutes after thrombus formation, the 50 mg edoxaban group significantly reduced thrombus volume compared to the placebo group, and promoted spontaneous thrombolysis.
At the same time, blood levels of t-PA, PAI-1, and TAFI, gene expression, and immunological expression were examined as activators and inhibitors of the fibrinolytic system, respectively. The results showed that the 50 mg edoxaban group significantly increased vascular and hepatic t-PA mRNA expression and decreased immunological expression of TAFI compared to the placebo group.
We reasoned that one possible explanation for these results could be the following. Regarding the elevated t-PA mRNA levels, we hypothesized that: F-Xa affects tPA expression as a result of intracellular signaling via binding to PAR1 and PAR2; edoxaban co-crystallizes with FXa in the X-ray structural analysis results indicate that the pyridine and oxalate moieties of edoxaban bind to the F-Xa S1 site and the tetrahydrothiazolopyridine moieties bind to the F-Xa S4 site [4,19], suggesting that edoxaban inhibits binding of F-Xa to PAR1 PAR2, which we reasoned that edoxaban may have inhibited the binding of F-Xa to PAR1 PAR2, resulting in an increase in t-PA expression levels. This requires specific verification.
Furthermore, F-Xa is known to be degraded by Plasmin to F-Xaβ, which enhances Plasminogen activation by t-PA [20,21].
F-Xaβ is further degraded and loses its action, but edoxaban may bind to the active site of F-Xa as described above, preventing its degradation by plasmin and maintaining the enhancement of fibrinolytic activity by t-PA with F-Xaβ. The decreased immunological expression of TAFI may be due to indirect inhibition of thrombin generation by edoxaban. The lack of changes in the genetic and immunological expression of PAI-1 could be due to the fact that PAI-1 is synthesized in cells and tissues such as liver, spleen, megakaryocytes, placenta, adipocytes, smooth muscle cells, macrophages, platelets, and vascular endothelial cells [22], and given that more than 90% of PAI-1 is contained in platelets [23,24], evaluation based solely on variation in mRNA expression of PAI-1 derived from blood vessels or liver may not be optimal. Since TAFI seems to be more effective than PAI-1 in inhibiting fibrinolysis [10], the effect of activated factor X inhibition on fibrinolytic activity may be more effective for TAFI than for PAI-1. Further analysis is needed.
Since thrombolysis is thought to occur when the dynamic balance between plasmin activity and plasmin inhibition exceeds plasmin activity, the increased mRNA levels of the fibrinolytic activator t-PA and the immunological decrease in TAFI may have shifted this fibrinolytic system dynamics toward increased fibrinolysis. This is the first paper to report that F-Xa-directed DOAC promotes thrombolysis in vivo model.
In summary, long-term treatment with edoxaban promoted thrombolysis in an in vivo thrombolysis model accompanied by increase mRNA levels of t-PA and decrease immunological expression of TAFI. In contrast, it had no effect on atherosclerosis development in Apoe-/- and Ldlr-/- DK mice.
These results provide evidence that edoxaban, an F-Xa-directed DOAC, enhance spontaneous thrombolytic activity and suggest a potential new additive effect of F-Xa-directed DOACs.
Oral direct F-Xa inhibitor, edoxaban (edoxaban tosilate hydrate) was kindly donated by Daiichi Sankyo Company Limited (Tokyo, Japan).
This research was supported by Daiichi Sankyo Company Limited (Tokyo, Japan). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors have no conflicts of interest directly relevant to the content of this article.