A natural sleeping pill and "elixir" (longevity-promoter) called "Melatonin" is one of the first natural anti-melanogenic compounds, and originally derived from bovine pineal glands. However, currently the majority of melatonin product in the market is chemically synthesized. Since then a wide variety of anti-melanogenic compounds such as curcumin, CAPE (Caffeic Acid Phenethyl Ester), and Artepillin C (ARC) were identified in nature as well as chemically synthesized compounds such as Gleevec. These anti-melanogenic compounds share a series of interesting biological properties such as anti-cancer, anti-inflammatory, anti-ageing (longevity-promoting), immune-boosting, anti-malaria, and anti-diabetic activities. However, the precise molecular mechanism underlying their anti-melanogenic action has remained to be clarified until recently. Finally we found the first clue to understanding of the "melanogenic" signalling pathway involving the major oncogenic/ageing kinase called PAK1 (RAC/CDC42-activated kinase 1) By siRNA-based silencing of PAK1 gene in murine melanoma cell line (B16F10): (i) The major growth hormone in serum called PDGF (Platelet-Derived Growth Factor) is essential for the inducible melanogenesis, but not for the exponential growth per se, and (ii) This cytokine activates its receptor called PDGFR, a Tyr-kinase on the cell surface, and induces another cell surface receptor Tyr-kinase called EGFR (Epidermal Growth Factor Receptor), to form a heterodimer with PDGFR, activating the down-stream melanogenic/oncogenic signalling pathway involving PAK1 and eventually MITF (Microphthalmia-Associated Transcription Factor) essential for expression of Tyrosinase and its Related Protein (TRP) genes. Melatonin and many other anti-melanogenic compounds block PAK1 directly or indirectly, and eventually down-regulate tyrosinase or TRP which is essential for melanin biosynthesis from an amino acid called tyrosine.
Melanogenesis, PAK1, PDGFR, EFGR, Melatonin, Propolis
It appears that there are at least two distinct signalling pathways that control melanogenesis: one is an "inducible" pathway, and the other is "basal" (non-inducible) one. The latter determines the color (black, yellow or white) of skin and hairs depending on distinct races, and it is genetically determined. The former inducible melanogenesis depends on UV irradiation and melanogenic hormones such as alpha-MSH (Melanocyte Stimulating Hormone), and is independent of races or genetic background [1,2]. Here we shall discuss mainly on the former (inducible melanogenesis), naturally because a variety of cosmetics containing anti-melanogenic compounds could be useful for so-called "skin-whitening/lightening" effect.
Melanogenesis (melanin biosynthesis) is a complex process starting hydroxylation of Tyrosine (Tyr) by tyrosinase in melanocytes or hair cells. Thus, a person who lacks tyrosinase gene or TRPs (Tyrosines-Related Proteins) is born as so-called "albino", having white hairs, white skin and red eyes, just like albino mice, white horses or kangaroos. A series of transcription factors control the expression of tyrosinase/TRPs gene. One of them is called MITF. The MITF gene is under the control of beta-catenin which is the direct substrate of the oncogenic/ageing kinase PAK1 [1].
A few years ago, we found that one of herbal PAK1-blockers called "Hispidin", a metabolite of DK (5,6-dehydrokawain), from leaves of Alpinia zerumbet in Okinawa reduced significantly alpha-MSH (Melanocyte Stimulating Hormone) - induced melanogenesis of murine melanoma cell line (B16F10) in cell culture [3]. One of reasons why we got interested in this subtropical plant was that the extract of Alpinia was found to be an elixir that extends the healthy lifespan of C. elegans [4]. Since we also found that PAK1-deficient mutant (RB689) of C. elegans lives 60% longer than the wild-type, clearly indicating that the major oncogenic kinase PAK1 shortens the healthy lifespan of this worm [5], we started identifying the PAK1-blockers including DK and its metabolite "Hispidin" in this extract.
To determine directly whether this anti-melanogenic activity of Hispidin is due to its anti-PAK1 activity or anything else, we silenced PAK1 gene in this cell line with PAK1-specific siRNA. As expected, PAK1-silencing clearly reduced the melanogenic activity of this cell line by around 50%, proving that PAK1 is essential for alpha-MSH-induced melanogenesis [1]. However, it became clear that the remaining 50% of melanogenesis is not affected by PAK1-silencing, although PAK1 expression level reached nearly null. Furthermore, a Korean group independently found that another member of PAK family kinases called PAK4, but not PAK2, is also involved in the melanogenesis of the same melanoma cell line [2]. Both PAK4 and PAK2 are abundantly expressed in this cell line, compared with a very low level of PAK1. Unfortunately, however, they concluded that PAK1 is not involved in melanogenesis of this cell line, because of the following three reasons: (i) They failed to detect PAK1 by western blot, (ii) Over-expression of the constitutively active mutant (T423E) of PAK1 does not boost the melanogenesis, and (iii) pPAK1 (Phosphorylation at Thr 423) is not detected even by alpha-MSH treatment [2].
Thus, we asked this Korean group to collaborate with us, confirming that over-expression of "native" (wild-type) PAK1 gene boosts the alpha-MSH-induced malanogenesis by 5-6 folds, but not the basal level of melanogenesis [1]. Thus, both of us reached to the conclusion that PAK1 is responsible mainly for the "inducible" melanogenesis, while PAK4 is responsible mainly for the remaining basal (non-inducible) melanogenesis. Furthermore, it should be pointed out once again that autophosphorylation of PAK1 at Thr 423 has nothing to do with the "inducible" melanogenesis, because over-expression of the so-called "constitutively active" mutant (T423E) of PAK1 has no effect on melanogenesis, and alpha-MSH has no effect on the phosphorylation of PAK1 at Thr423 [2]. In other words, the popular "sloppy" (easy-going) western blot analysis of both PAK1 protein and pPAK1 levels often tends to mislead the people to a wrong conclusion. Clearly, a very "minute" amount of PAK1 is sufficient for controlling the inducible melanogenesis by a robust phosphorylation of PAK1 at as yet "unknown" site(s). Thus, we should urge all PAK1-researchers to assay its kinase activity more directly in cells, by using either gamma-32P-ATP or "Macaroni-Western" (IP) Glo-kinase assay kit [1], instead of western blot. For at least three distinct Tyr-kinases (ETK, FYN and JAK2) are involved in full activation of PAK1 in cells, in addition to RAC/CDC42 and PIX, and these Tyr-kinases have nothing to do with Thr423 phosphorylation of PAK1 [6].
More interestingly, we found that alpha-MSH-induced melanogenesis of B16F10 cells requires the 10% bovine fetal serum [1], although the serum is not essential for their exponential growth per se. However, the serum is definitely required for their survival before their attachment to the substratum, clearly indicating that their "anchorage-independent" growth or survival requires serum [1]. Actually serum alone without alpha-MST still can stimulate the inducible melanogenesis by around 50%, and the combination of both serum and alpha-MSH fully promotes the inducible melanogenesis by almost 100% [1].
More than a decade ago we found that PDGF in serum is the major growth factor that activates PAK1 in other cells in a RAS-dependent manner, and the PDGF-dependent activation of PAK1 requires not only PDGF Receptor (PDGFR), but also EGFR (EGF Receptor) [7]. Either PDGFR-specific inhibitor (AG 1295) or EGFR-specific inhibitor (AG 1478) alone inhibits completely the serum/PDGF-induced activation of PAK1 [7]. It is most likely that the PDGFR-EGFR heterodimerization is required for the PDGF-induced PAK1-activation. Thus, we currently speculate that serum-induced activation of PAK1 leads to 50% increase in inducible melanogenesis, and the combination of serum and alpha-MSH leads to 100% increase in inducible melanogenesis by a robust activation of PAK1. In fact, although alpha-MSH alone is unable to activate PAK1, serum alone activates PAK1 by 2 fold, and the combination of both serum and alpha-MSH activates PAK1 by 4-5 folds [1]. Independently a Japanese group confirmed that PDGF induces alpha-MSH-dependent melanogenesis in human melanocytes [8].
Why does alpha-MSH require serum/PDGF for the activation of PAK1? The following is our working hypothesis (for detail, see Figure 1). First of all, alpha-MSH activates PAK1 through a Tyr-kinase called JAK2. However, RAS is also required for JAK2 activation. To activate RAS, the cell surface PDGFR-EGFR signalling complex is required. This pathway activates not only alpha-MSH-JAK2 pathway via RAS, but also RAS-RAC pathway which eventually activates PAK1. Thus, alpha-MSH alone cannot activate PAK1. However, the PDGF-induced activation of PDGFR-EGFR complex leads to the activation of RAS, and in turn the alpha-MSH-dependent activation of JAK2, leading to PAK1 activation. Thus, in theory, any compounds that interfere with this melanogenic signalling pathway could be anti-melanogenic.
 Figure 1: PDGF/µ-MSH-dependent melanogenic signaling pathway. View Figure 1
Figure 1: PDGF/µ-MSH-dependent melanogenic signaling pathway. View Figure 1
In this brief review we shall introduce a dozen of PAK1-blocking anti-melanogenic compounds (natural or synthetic), which could be useful as ingredients of so-called "skin-lightening" cosmetics which would cause no side effect. These PAK1-blockers could be the better alternatives for the conventional "direct tyrosinase-inhibitors" that often cause the so-called "white-spotting" irreversible side effect. Regarding the biological roles of melanocytes and a few other skin cells in general, we (molecular oncologists) would urge readers to study an expert review on this subject by dermatologists.
Melatonin is a pineal hormone in brain that was originally identified as an anti-melanogenic agent by Aaron Lerner's group at Yale University in 1958 [9]. It is biosynthesized from serotonin (See Figure 2). Later, it was found to be a natural circadian sleeping pill that is daily synthesized/secreted only when light is off or sun is set. According to 1995 book by Russel Reiter and Jo Robinson [9], Melatonin is a multi-functional hormone or "wonder drug" that is useful for therapy of a wide variety of diseases/disorders including cancer, AD (Alzheimer's disease), diabetes (type 2), hypertension, inflammatory diseases, depression, schizophrenia, epilepsy, infectious diseases such as HIV, and so forth, all of which are known to be PAK1-dependent [6]. Most interestingly, it was found by Georges Maestroni's team in Switzerland around 1985 to be a powerful elixir that extends the healthy lifespan of 19-months-old mice by 20% as well as Drosophila and rats [10]. To the best of our knowledge, melatonin is the very first among natural or synthetic compounds to be identified as a "real" elixir (longevity-promoter or anti-ageing drug). Until then the "elixir for eternal youth" remained to be a "fictional"/dream compound that appears only in fairy tales or Greek mythology. Furthermore, the same Swiss group found around 1995 that like propolis, melatonin boosts the immune system, in particular helper T-cells [8]. Regarding the molecular mechanism underlying these therapeutic effects, it is most likely that melatonin is a PAK1-blocker. Most notably, melatonin activates the anti-oncogenic kinase AMPK [11]. Since most of natural AMPK activators such as propolis and curcumin block the oncogenic/ageing kinase PAK1 without any exception [6], it is almost certain that melatonin also blocks PAK1, probably through the longevity kinase LKB1 that inactivates PAK1, and activates AMPK, simultaneously [6]. We are currently confirming this notion directly. Furthermore, since IC50 of melatonin against the growth of A549 cancer cells in cell culture is rather high (around 1.3 mM), we are trying to potentiate its anti-cancer/anti-melanogenic activity by replacing acetyl moiety with the basic amino acids such as Arg, in an attempt to boost its cell-permeability.
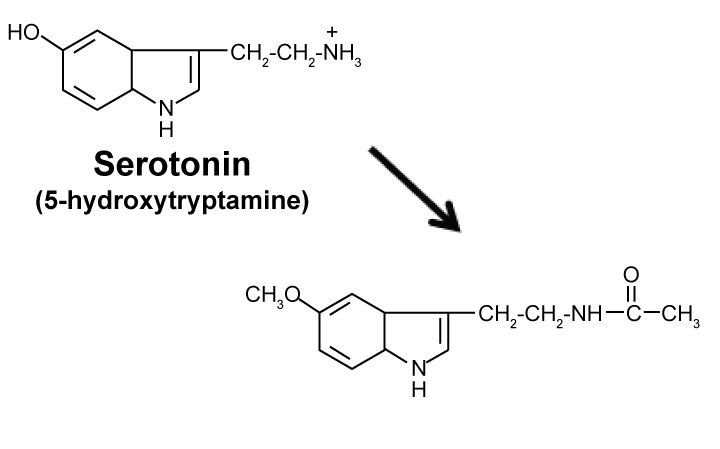 Figure 2: Biosynthesis of melatonin (anti-Melanogenic metabolite of Serotonin). View Figure 2
Figure 2: Biosynthesis of melatonin (anti-Melanogenic metabolite of Serotonin). View Figure 2
Propolis, an ethanol extract of honeybee comb, has been used as a traditional medicine since the ancient Egyptian era. It is useful for wound healing and among powerful antibiotics killing both bacteria and viruses [6]. In addition, it was commonly used for preparing mummies of deceased royal families stored under pyramids. Hippocrates, the Father of Medicine, in ancient Greece, coined this antibiotic from bee comb "Pro-polis" (Pro for protection, and Polis for city or comb). In 1960s, propolis was rediscovered as an herbal medicine for the treatment of influenza virus and other infectious/inflammatory diseases [6]. In late 1980s, propolis was found to inhibit the growth of cancer cells, and one of its anti-cancer ingredients was identified CAPE (Caffeic Acid Phenethyl Ester) [12]. The plant sources of propolis widely differ, depending on areas where propolis is harvested. CAPE-based propolis such as Bio 30 from New Zealand is derived from young buds of poplar trees and willow. However, Artepillin C (ARC)-based propolis from Brazillian Green Propolis (GP) is derived from Baccharis dracunculifolia [6]. Subtropical propolis from Okinawa, Taiwan, and Hawaii is based on Nymphaeols, geranylated flavonoids, which are derived from young buds of Macaranga tanarius [13]. However, all propolis products appear to share the common biological property that is to block the oncogenic/ageing kinase PAK1 somehow, eventually extending the healthy lifespan of C. elegans at least [6, 13]. Most importantly, Bio 30 is the most potent among propolis products to inhibit the growth of various cancers including Gemcitabine-resistant terminal pancreatic cancer, and suppress even brain tumors such as NF tumors in clinical trials, clearly indicating that its anti-PAK1 ingredients pass effectively the Blood Brain Barrier (BBB) [6].
Among the herbal PAK1-blockers, apigenin from the CAPE-based propolis or camomile flowers is the first that has been proven to extend the lifespan of nematode (C. elegans) in a FOXO-dependent manner [14]. Apigenin is a flavonoid that blocks PAK1 by directly down-regulating PI-3 kinase [15]. However, since apigenin is structurally related to the direct PAK1-inhibitors called nymphaeols, but lacks their geranyl side chains which appear to be non-essential for their anti-PAK1 activity per se, it is quite possible that apigenin and several other flavonoids such as naringenin and sakuranetin also directly inhibit PAK1, and currently we are testing such a possibility.
Caffeic Acid (CA) from propolis was also shown to extend the lifespan of this worm in a FOXO-dependent manner [16]. CA directly down-regulates RAC, leading to inactivation of PAK1 [17].
CAPE (Caffeic Acid Phenethyl Ester) is a natural ester of CA with phenethyl alcohol, and among the major PAK1-blockers in propolis. CAPE is the first propolis ingredient whose anti-cancer activity was identified [12]. The anti-cancer activity of CAPE in cell culture (IC50 = 10 μM) is 10 times higher than CA (IC50 = 100 μM) [18]. Like CA, CAPE down-regulates RAC, leading to the inactivation of its effector PAK1. Interestingly, CAPE at 100 μM extends the lifespan of C. elegans by around 15% [19], as we have proven that CAPE reduces the brood size and activates HSP16 gene through FOXO in this worm [5], clearly indicating that CAPE is also a so-called "elixir" (meaning longevity promoter) as well as a herbal anti-cancer agent that is clearly more potent than CA. As described later in detail, we have recently synthesized water-soluble and highly cell-permeable triazolyl ester of CA called 15C, whose anti-cancer activity is over 400 fold more potent than CA [18]. Among propolis products, so far a CAPE-based propolis called Bio 30 from New Zealand is the most potent, and inhibits the growth of A549 lung cancer cells with IC50 around 8 μg/ml, and melanogenesis of B16F10 melanoma by down-regulating the tyrosinase level with IC50 around 6 μg/ml (see Figure 3).
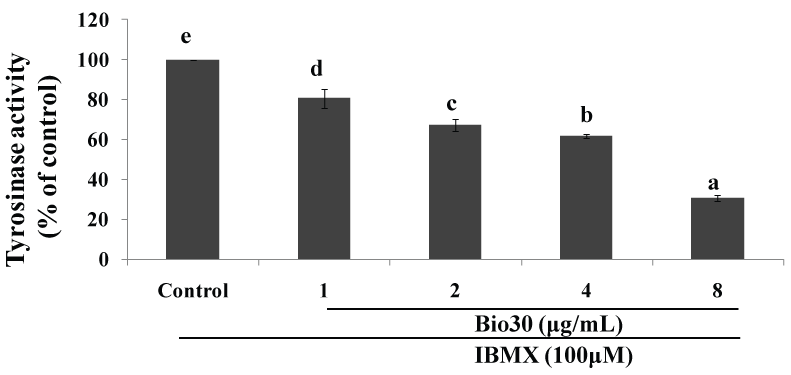 Figure 3: Down-regulation of tyrosinase level by bio 30 (CAPE-based propolis).
Figure 3: Down-regulation of tyrosinase level by bio 30 (CAPE-based propolis).
2 × 104 cells (B16F10) were seeded for 24 hrs, and then in the presence of IBMX treated with Bio 30 at the indicated concentrations for 72 hrs. L-Dopa was added as a substrate to cell lysates for perfoming the tyrosinase assay. Tyrosinase level was measured at 490 nm (Be-Tu PT, et al., unpublished observation).
View Figure 3
Artepillin C (ARC) is the major anti-cancer ingredient of Brazillian Green Propolis (GP), and blocks PAK1 in cell culture (IC50 = 25 μM) [18]. Like CAPE, ARC reduces the brood size and activates HSP16 gene in a FOXO-dependent manner in C. elegans [5]. Thus, it is most likely that ARC is also an elixir. Like CA, ARC bears COOH moiety that hampers its penetration across the negatively charged plasma membranes. Thus, we recently boosted its anti-cancer activity mainly by boosting its cell-permeability by 100 fold through triazolyl esterization without any loss of its water-solubility [18]. To the best of our knowledge, ARC is the first propolis ingredient that has been proven to promote the growth of hair cells in cell culture with the Effective Dose (ED) around 20 μM [13], confirming that (i) ARC is the major contributor to the anti-alopecia effect of Brazillian green propolis previously demonstrated in vivo (a mouse model), and (ii) PAK1 normally suppresses the hair growth or promotes a hair loss (alopecia).
Propolis from subtropical Pacific regions such as Okinawa, Taiwan and Hawaii is very distinct from those found in Brazil or New Zealand. It contains a series of geranylated flavonoids such as Nymphaeols A-C [13]. In 2016, we found that Okinawa Propolis (OP) as well as its ingredients Nymphaeols A-C directly inhibit PAK1 in vitro, and suppress the growth of A549 lung cancer cells with IC50 around 12 μg/ml [20]. Furthermore, we found that OP extends the healthy lifespan of C. elegans at 1 μg/ml [13], and inhibits melanogenesis of B16F10 melanoma by down-regulating their tyrosinase level with IC50 around 30 μg/ml [13]. Thus, it is most likely that Nymphaeols are among herbal anti-melanogenic agents, as other propolis ingredients such as apigenin and CAPE.
Curcumin directly inhibits PAK1 in vitro with IC50 around 16 μM [20], and the growth of a variety of cancer cells such as A549 with IC50 around 23 μM [20]. Curcumin also has been shown to extend the lifespan of nematode [21]. Thus, it would be worth testing if curcumin also inhibits melanogenesis.
The major problem/setback for clinical application of curcumin is its poor bioavailability (water-insolubility). A decade ago, an oncologist group led by Razelle Kurzrock at MD Anderson Cancer Centre in Texas successfully potentiated the bioavailability of curcumin by liposomes for therapy of pancreatic/colon cancers [6], and according to their 2008 clinical trial report, this "liposome" recipe appears to work in clinical trials (phase II) for some advanced pancreatic cancer patients [6].
Back in 1928, Joanna Brandt, an US immigrant from South Africa published a small booklet entitled "Grape Cure" for cancer, describing how she managed to cure her own stomach cancer by eating mainly red grapes [22]. Since then, many oncologists tried to find the molecular mechanism underlying her "grape therapy of cancers".
Eventually 7 decades later, in 1997, John Pezzuto's team at University of Illinois in Chicago found that a polyphenol called resveratrol (R3) is the major anti-cancer ingredient in red grapes [23]. More interestingly, they recognized that R3 inhibits COX-2 directly in vitro as well as inactivates COX-2 gene (with IC50 = 5 μM), showing an anti-inflammatory effect as well [24]. Thus, these finding suggest that red wines would be more beneficiary than white wines (which lacks R3) for our good health in general. In 2003, David Sinclaire at Harvard Medical School claimed that R3 directly activates the longevity gene product called Sirtuin (SIRT) in yeast and extends the lifespan of yeast [25]. Around 2007, R3 was found to be an elixir that extends the lifespan of C. elegans [26]. How does the down-regulation of COX-2 by R3 contribute to the extension of the lifespan? PAK1 is essential for the activation of COX-2 gene [27]. Thus, there is a possibility that R3 somehow blocks PAK1, leading to the down-regulation of COX-2. During 2014-2015, we confirmed that R3 directly inhibits PAK1 in vitro with IC50 = 15 μM, and identified a variety of more potent herbal PAK1-inhibitors such as FRA (Frondoside A) from sea cucumber and Nymphaeols from Okinawa propolis [20,28]. Finally, in 2014, a Korean group found that R3 indeed inhibits UV-induced melanogenesis in guinea pig skin [29].
Hispidin and its derivatives such as DK (5, 6-dehydrokawain) (see Figure 4, top) are widely distributed in a variety of edible mushrooms. Very fascinatingly, these mushrooms such as Neonothopanus gardneri from Brazil's palm forests glow softly green in the dark (see Figure 4, bottom), mainly due to the bright "Hispidin-based" biofluorescence/luminescence:
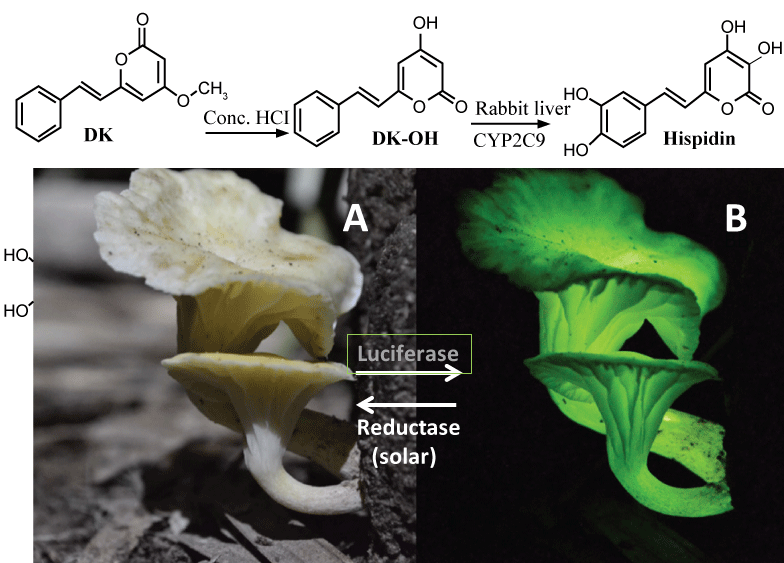 Figure 4: DK and its metabolite (Hispidin & Luciferin). Top, synthesis of hispidin from DK. Bottom, 3-OH hispidin-based fluorescence of a Brazilian mushroom: (A) Daytime (by "solar" recyclase); (B) At night (by luciferase = oxidase). View Figure 4
Figure 4: DK and its metabolite (Hispidin & Luciferin). Top, synthesis of hispidin from DK. Bottom, 3-OH hispidin-based fluorescence of a Brazilian mushroom: (A) Daytime (by "solar" recyclase); (B) At night (by luciferase = oxidase). View Figure 4
https://www.sciencenews.org/blog/science-ticker/how-mushroom-gets-its-glow
According to an international team led by Prof. Cassius Stevani in Brazil, this fluorescence (LED) depends on a unique ATP-independent oxidase/decarboxylase called "luciferase" and its substrate (luciferin) called "3-hydroxyl hispidin" (3-OH hispidin) [30]. Finally, after sunset, the oxidized form of 3-OH hispidin glows. Furthermore, just like melatonin synthesis in pineal glands during night [9], this bio-LED system is controlled by a "circadian" clock consisting of (a) "night-time" luciferase and (b) "solar/temperature-dependent" reductase/recyclase), recycling the luciferin during daytime, and releasing the green biofluerescence only at night, to attract a variety of insects in forest (see Figure 4, bottom).
Thus, in theory, this mushroom reductase system could be used a solar bio-battery accumulating the luciferin as a new "green" energy source for illumination.
Hispidin itself is more cytotoxic to cancer cell lines such as A549 than any normal cells. Shortly after we found that an extract from leaves of Alpinia zerumbet in Okinawa extends the healthy lifespan of C. elegans [4], we identified hispidin, a metabolite of DK, among the anti-oncogenic ingredients of this extract, and found that hispidin directly inhibits the oncogenic/ageing kinase PAK1 in test tube with IC50 around 6 μM [28]. Since PAK1 is essential not only for cancer growth, but also for PDGF-dependent melanogenesis of B16F10 melanoma cells [1], we tested its potential anti-melanogenic activity in cell culture. Hispidin inhibits the melanogenesis by down-regulating intracellular tyrosinase level by 60% at 20 μg/ml (around 80 μM) without any effect on the "exponential" growth of melanoma cells, clearly proving again that their inducible melanogenesis is PAK1-dependent, while their growth is PAK1-independent [3]. These observations suggest that the cell-permeability of the herbal PAK1-inhibitor "hispidin" is rather poor, although it inhibits both "inducible" melanogenesis of melanoma and the growth of A549 cancer cells with IC50 around 25 μM [1,13, 28]. Thus, we synthesized a series of hispidin derivatives (H1-H3) which are clearly more potent than hispidin [28].
Furthermore, our structure-function analysis of these hispidin derivatives [28] strongly suggests, if not proven as yet, that 3-OH hispidin also directly inhibits/binds PAK1. In other words, there is a possibility that we could label PAK1 in living cells with this bio-LED. We are currently testing if 3-OH hispidin could distinguish between "active" (monomer) and "inactive" (homo-dimer) forms of PAK1 in vitro. If it does, we could monitor the changes in kinase activity of PAK1 in living cells with this fluorescent probe at concentrations far below the IC50.
Shikonin (see Figure 5), a natural naphthoquinone-based red pigment first isolated in 1977 by a Japanese group led by Prof. Ushio Sankawa at Tokyo University, from red roots called "Shikon" (meaning "violet root" in Japanese) or the traditional Chinese medicine Zi Cao (gromwell), has been reported to possess both anti-cancer and anti-inflammatory activities [31]. Results from a clinical study using a shikonin-containing mixture demonstrated its safety and efficacy for the treatment of late-stage lung cancer. Later, shikonin was shown to be effective for treatment of AIDS and skin-whitening as well.
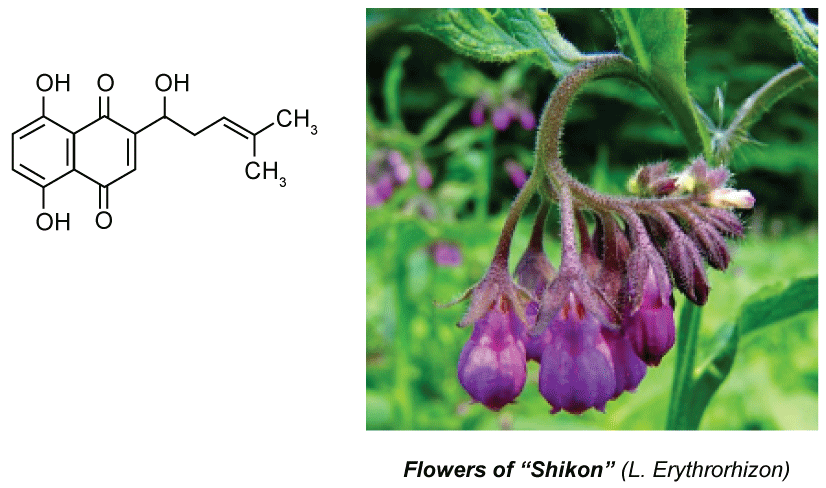 Figure 5: Shikonin, red pigment from "Shikon" roots. View Figure 5
Figure 5: Shikonin, red pigment from "Shikon" roots. View Figure 5
However, the mechanism underlying this pharmaceutical activity of shikonin remained to be clarified till recently. The first clue was found by a Takashi Kondo's team at Toyama University. Shikonin induces HSPs (heat shock proteins) [32]. Since HSP genes are suppressed by PAK1, a possibility has raised that shikon is a PAK1-blocker. In support of this notion, Shikonin was found to suppress PAK1-dependent angiogenesis by inhibiting a Tyr-kinase called JAK2 directly at 100 nM [33], which is essential for PAK1 activation. Recently mass production of shikonin by a tank callus culture of "Shikon" (Lithospermum erythrorhizon) has been successfully done by Mitsui Petrochemicals in Japan for production of a shikonin-based lip-stick and other cosmetics [34].
Glaucarubinone is a triterpenoid/quassinoid (see Figure 6) derived from a bitter tree (Simaroubaceae family) in Amazon forest. The extract of this tree bark has been used as a traditional medicine by local Amazon people for treatment of a variety of diseases including malaria infection which is now proven to be PAK1-dependent. Around 1981, its major active ingredient was identified as glaucarubinone, and rather surprisingly, it was found to show a potent anti-cancer activity [35]. Around 1993, Paul Grieco at Indiana University successfully managed to synthesize this compound chemically and patented it [36]. However, the molecular mechanism underlying its anti-cancer activity remained to be clarified until recently. In 2009, John Beutler's group at NCI-Frederick found the first clue: this compound inhibits the function of an oncogenic transcription factor called AP-1 in cancer cells with IC50 around 20 nM [37]. Since AP-1 is downstream of PAK1, we started suspecting that it might be a PAK1-blocker. In collaboration with his team, we confirmed that glaucarubinone indeed blocks PAK1 in cell culture, and another group led by Hong He at Melbourne University Hospital found that it blocks both PAK1 and PAK4 in vivo, inhibiting the growth of human pancreatic and colon cancer xenograft in mice at 1-2 mg/kg, i.p. twice a week [38]. To the best of our knowledge, this compound is the most potent anti-cancer agent among herbal PAK1-blockers. Since melanogenesis also depends on both PAK1 (inducible) and PAK4 (basal), we have tested its anti-melanogenic effect using B16F10 melanoma cells [39], and confirmed that it indeed inhibits the melanogenesis with IC50 around 440 nM (Be-Tu PT, et al., unpublished observation). Interestingly, in 2011, a group at Jena University in Germany found that this compound extends the lifespan of C. elegans, suggesting that it would not cause any side effect during cancer therapy or cosmetic skin-treatment [40].
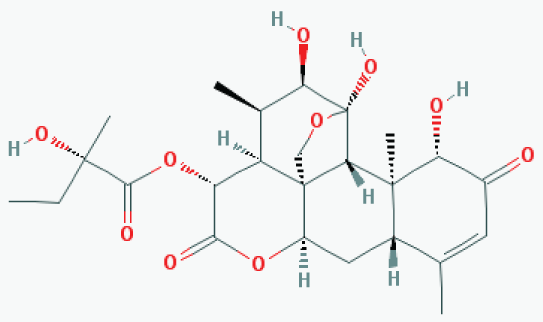 Figure 6: Glaucarubinone. View Figure 6
Figure 6: Glaucarubinone. View Figure 6
Although so far there exists no direct evidence for proving that FK228 inhibits melano-genesis, there is a very strong reason to suspect that this potent HDAC inhibitor would be the most potent anti-melanogenic agent available on the market. More than a decade ago, we found that FK228 blocks PAK1 in cell culture with IC50 below 1 nM and inhibits completely the growth of Tamoxifen-resistant breast cancer in vivo with 2.5 mg/kg, i.p. twice a week [41]. Interestingly, just a year later, a Japanese group at Jichi University found that FK228 inhibit the PAK1-dependent growth of human malignant melaloma cells carrying an oncogenic B-RAf mutant by blocking the oncogenic RAS-PI3 kinase signalling pathway which is upstream of PAK1 [42]. Thus, they missed the very good chance to look at the potential anti-melanogenic effect of FK228. If they have tested the effect of FK228 on the melanogenesis of B16F10 melanoma cells instead, they could have discovered its anti-melanogenic activity straight away, simply because the exponential growth of this mouse melanoma cell line is independent of PAK1.
FK228 is a water-soluble cyclic peptide from a soil bacterium, and was originally discovered as a potent anti-RAS cancer agent in 1994 by Fujisawa Pharma [43]. Later it was rediscovered to inhibit directly an oncogenic enzyme called HDAC with IC50 around 1 nM [43]. Shortly after we discovered that FK228 blocks PAK1, we tried to develop FK228 for a therapeutic of formidable pancreatic cancers and brain tumors such as NF (neurofibromatosis). Unfortunately, however, we found that FK228 fails to pass the BBB (Blood Brain Barrier), and tends to induce a strong "drug-resistance" in pancreatic cancers. Thus, we gave up FK228 after all. Finally, around 2009, FK228 was approved by FDA as the first therapeutic only for a rare malignant solid tumor called CTCL (Cutaneous T-Cell Lymphoma), and marketed under the brand name "Istodax" by CelGene [44]. Thus, in principle, Isodax could be used as a potent skin-lightening cosmetic agent as well.
If we understand correctly, Gleevec is one of the first synthetic PDGFR-inhibitors that show the clear anti-melanogenic activity in human patients suffering from CML, GIST etc. Originally Gleevec was developed by Novartis (then called Ciba-Geigy) toward the end of 1990s as an ABL-specific inhibitor for treatment of Chronic Myelogenous Leukemia (CML) and Acute Lymphocytic Leukemia (ALL) that is Philadelphia Chromosome-Positive (Ph+) as well as certain types of Gastrointestinal Stromal Tumor (GIST). However, it turns out to inhibit two other Tyr-kinases, PDGFR (PDGF Receptor) and KIT as well. During prolonged treatment with Gleevec, some patients noticed that their skin or hair color becomes clearly lighter than used to be, indicating its anti-melanogenic (side) effect. This is not a big surprise, however, because the inducible-melanogenesis depends on PDGF in serum that activates its receptor PDGFR, eventually activating the melanogenic kinase PAK1 as well [1]. According to a 2014 report by a Chinese group, Gleevec indeed inhibits melanogenesis of Human Epithelial Monocytes (HEM) by inhibiting both PDGFR and KIT [45].
So-called PAK1/PAK4-specific inhibitor called PF3758309 (PF309 in short) was developed in 2010 by a group led by Bill Murray at Pfizer Oncology in US, and inhibits both PAK1 and PAK4 with IC50 around 10 nM in test tube [46]. PF309 has been shown to inhibit several human cancer cell lines in cell culture with IC50 ranging 10-20 nM [46]. However, its in vivo test was not so successful (in particular for clinical trials), mainly because it causes a serious internal bleeding if we understand correctly. PAK4-deficinecy leads to embryo lethality in mice, mainly due to cardiovascular failure, while PAK1-deficiency improves significantly the health conditions [6]. Interestingly, however, this inhibitor was found by a Korean group led by Eung-Gook Kim to inhibit the melano-genesis of B16F10 melanoma and Human Epithelial Melanocyte (HEM) in cell culture with IC50 ranging 30-60 nM, and also suppresses significantly UV-induced malanogenesis in mouse skin with IC50 around 2 µM [2]. Thus, instead of an anti-cancer reagent, it could be used as a skin-whitening cosmetic agent.
Both ARC and CA are among propolis ingredients that block the oncogenic kinase PAK1 and inhibit the growth of solid tumors [6]. However, due to their COOH moiety, their cell-permeability is very poor. Thus, around 2015, we have successfully esterized both ARC and CA with the water-soluble 1,2,3-triazolyl alcohol via a simple chemical reaction called "Click Chemistry" (see Figure 7). This esterization boosted both anti-cancer and anti-PAK1 activities of ARC and CA by 100 and over 400 times, respectively, mainly due to a robust increase in their cell-permeability [18]. We also confirmed the anti-melanogenic activity of 15A (1,2,3-triazolyl ester of ARC) at least in B16F10 melanoma cell culture with IC50 around 0.6 µM (see Figure 8), 20 times more potent than ARC with IC50 around 12 µM [39]. This 15A-induced reduction in melanin content is due to a significant drop in the tyrosinase level in melanoma caused by the 15A treatment (see Figure 9). However, this assay is rather tricky, mainly because 15A and any other 1,2,3-triazolyl esters are converted to a pigment, whose chemical structure has not been determined as yet, presumably by mitochondrial reductase (or other related enzymes), as is MTT reagent which is a related "tetrazole" compound. Nevertheless, we believe both 15A and 15C (1,2,3-triazolyl ester) would be among the new powerful synthetic skin-whitening agents.
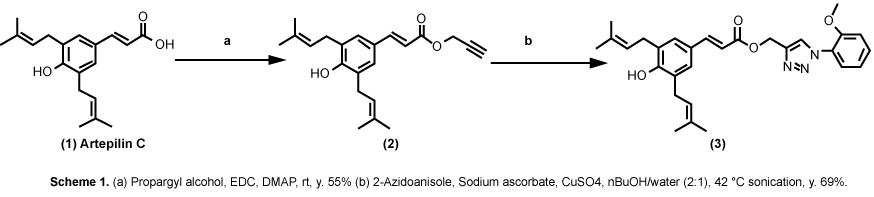 Figure 7: Click chemistry turning artepillin C (ARC) to "15A". View Figure 7
Figure 7: Click chemistry turning artepillin C (ARC) to "15A". View Figure 7
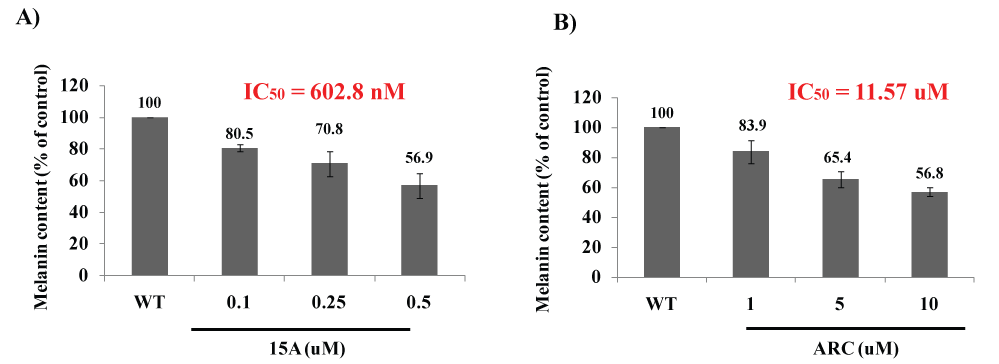 Figure 8: Anti-melanogenic activity of ARC and its 1,2,3-triazolyl ester (15A).
Figure 8: Anti-melanogenic activity of ARC and its 1,2,3-triazolyl ester (15A).
2 × 104 cells (B16F10) were seeded for 24 hrs, and then in the presence of IBMX treated with either 15 K (A) or ARC (B) at the indicated concentrations for 72 hrs. The melanin content was measured at 405 nm. 15A reduced the melanin content in cells with IC50 around 0.6 µM, while ARC did with IC50 around 12 µM (Be-Tu PT, et al., unpublished observation).
View Figure 8
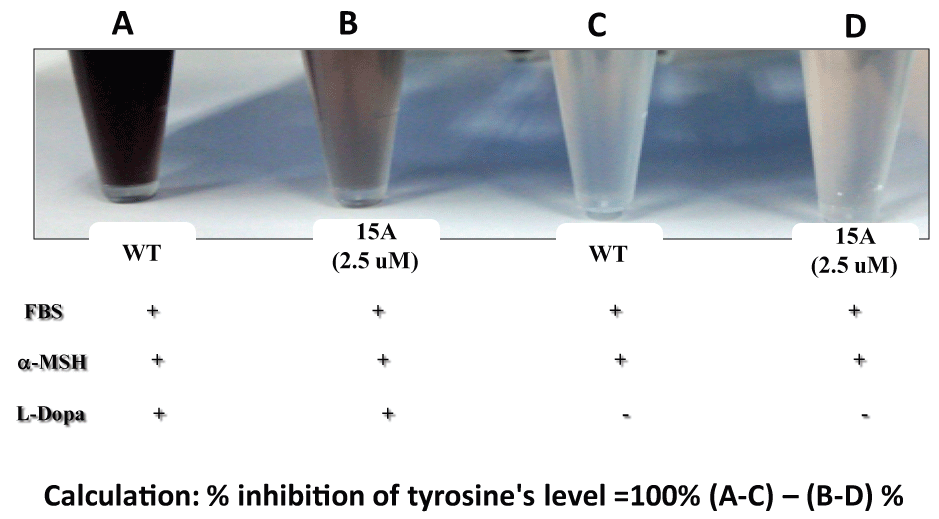 Figure 9: Suppression of tyrosinase levels by 15A.
Figure 9: Suppression of tyrosinase levels by 15A.
2 × 104 cells (B16F10) were seeded for 24 hrs, and then in the presence of both FBS and µ-MSH, treated with 15A (2.5 µM) for 72 hrs. For tyrosinase assay, L-Dopa was added to each cell lysate. Tyrosinase level was measured at 490 nm (Be Tu PT, et al., unpublished observation). Since 15A alone without L-Dopa (D) showed a significantly higher colored background, compared with the WT (C), due to "pigmentation" of 15A presumably by mitochondrial reductase or other related enzymes (Be-Tu PT, et al., unpublished observation).
View Figure 9
An old synthetic pain-killer called Ketorolac is another COOH-bearing PAK1-blocker which was sold by Roche. Ketorolac is a racemic mixture of R and S forms. R-form directly inhibits RAC, an activator of PAK1, while S-form inhibits directly COX-2, which is down-stream of PAK1 [47]. In order to boost its cell-permeability, we have done 1,2,3-triazolyl esterization of Ketorolac via Click Chemistry, and the resultant ester called 15 K is over 500-5000 times more potent than Ketorolac as the anti-cancer agent and anti-PAK1-blockers [47]. Interestingly, however, 15 K strongly inhibits the growth of B16F10 melanoma cell line with IC50 around 5 nM, which is not PAK1-dependent [47]. Thus, it appears that 15 K targets (in addition to RAC and COX-2) a third protein called Melanoma Oncogenic Factor (MOF) in this melanoma cells whose precise chemical nature still remains to be clarified. Since it inhibits the exponential growth of melanoma per se, it is still unclear whether 15 K is anti-melanogenic or not. In the future, we shall test its potential anti-melanogenic effect on human melanocytes.
Retinoic Acid (RA) is known to have an anti-melanogenic activity. However, since it causes an inflammatory action on skin, it is not suitable as a skin-whitening cosmetic agent. In 2014, during RA research, Noriko Takahashi's group at Hoshi University of Pharmacy in Tokyo recently found a synthetic new anti-melanogenic (but not inflammatory) agent called p-Decylamino Phenol (pDAP in short) which inhibits melanogenesis by 40% at 1 µM [48]. Its anti-melanogenic action is clearly stronger than a mushroom anti-melanogenic agent called Kojic Acid. Furthermore, the mechanism underlying its anti-melanogenic action is different from that of Kojic Acid.
Kojic acid inhibits directly (mushroom) tyrosinase in test tube, which is essential for melanogenesis. However, pDAP does not inhibit this enzyme directly. Instead, pDAP inhibits a melanogenic/oncogenic transcription factor called MITF which is essential for the expression of tyrosinase gene. Furthermore, pDAP selectively inhibits the MEK-ERK signaling pathway (with IC50 around 0.3 µM), downstream of PAK1, and not AKT [48]. Thus, it is most likely that pDAP blocks PAK1 somehow. It should be worth noting that pDAP does not inhibit the melanogenesis by more than 50%, suggesting that it does not block PAK4 which is responsible mainly for the basal melanogenesis. Thus, pDAP could be useful not only for skin-whitening, but also for cancer therapy and even promoting the longevity.
Around 2007, a group at Astellas Pharma in Japan developed a potent surviving-suppressor called YM-155. This compound (see Figure 10) suppresses almost completely the promoter function of survivin gene [49], instead of inhibiting directly the biological function of survivin per se. We shall very briefly describe what is the biological function of a protein called "survivin". A protease inhibitor called "Survivin" was found in 1997 by a group at Yale University, which blocks programmed cell death, either caspase-dependent apoptosis or ATG-dependent autophagy, of malignant cells, thus mainly promoting their survival [50]. Since survivin is highly expressed in malignant cells, but not in normal cells, survivin could be a selective target for anti-cancer drugs.
Interestingly, in 2013 a group at Indiana University found that PAK1-deficiency in mice inactivates survivin gene [51], clearly indicating that PAK1 is essential for transcription of survivin gene. Furthermore, we found that PAK1-deficient mutant of nematode (RB689) lives 60% longer than the wild-type [5], strongly suggesting the possibility that YM-155 could also extend the healthy lifespan of small animals such as nematode and mice by down-regulating survivin gene.
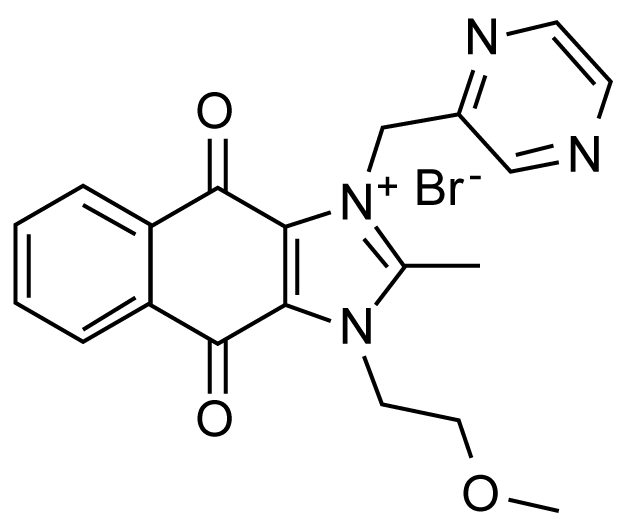 Figure 10: YM155 that blocks EGFR-PAK1-survivin signaling pathway. View Figure 10
Figure 10: YM155 that blocks EGFR-PAK1-survivin signaling pathway. View Figure 10
YM-155 inhibits the growth of several cancer cells with the IC50 ranging 10-100 nM, and the growth of PC3 prostate cancer xenograft in mice with daily dose (3 mg/kg) by more than 50% [49]. Since YM-155 suppresses the PAK1-dependent expression of COX-2 gene essential for survivin expression [52], there is a possibility that YM-155 might block PAK1 somehow. Recently, we confirmed the anti-PAK1 activity of YM-155 in A549 cancer cell culture (IC50 around 500 nM), an elixir activity in C. elegans [53], and its anti-angiogenic effect in HUVECs culture (IC50 around 25 nM) and in ovo (fertilized eggs, IC50 = 0.5 nmol/egg) as well [54]. YM-155 is currently in clinical trials (phase 2) for a variety of cancers including pancreatic cancer. Thus, we have a very good reason to predict that like FK228, YM155 also could serve as a potent skin-lightening cosmetic agent in the future.
As summarised in Figure 11, there are at least two distinct ways to block melanogenesis. The conventional way is to use tyrosine/L-Dopa antagonists for blocking the melanogenesis by inhibiting tyrosinase directly. These direct "tyrosinase-inhibitors" such as rhododenol and kojic acid sometimes tend to cause a serious (irreversible) "white-spotting" (leukoderma) on skin by their metabolites which selectively kill melanocytes. Thus, an alternative (far more sophiscated) approach, which belongs to a new generation cosmetic technology, is to block the melanogenic PDGF-PAK1-MITF signalling pathway (for detail, see Figure 1), leading to suppression of tyrosinase gene expression. This approach can be done by a series of PAK1-blockers such as CAPE and shikonin which do not affect the growth of any normal cells including melanocytes, and therefore cause no side effect. Furthermore, PAK1-blockers in general show anti-oncogenic, anti-inflammatory and even anti-ageing (longevity-promoting) properties as well. Thus, "PAK1-blockers"-based cosmetics would be far better than the conventional "tyrosinase-inhibitors"-based cosmetics.
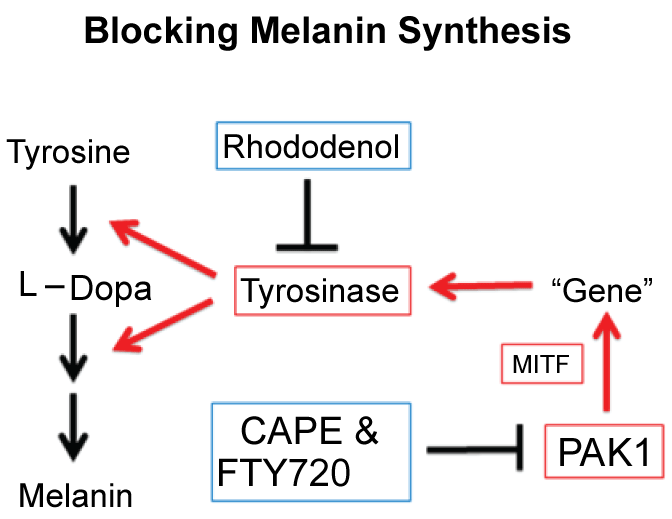 Figure 11: Two distinct ways to block melanogenesis. View Figure 11
Figure 11: Two distinct ways to block melanogenesis. View Figure 11
We are very grateful to Dr. Binh Ngyuen for his unpublished data on 15 K and YM155.