We report a rare case of trilateral retinoblastoma with uniocular involvement and spinal metastases.
Trilateral retinoblastoma is a syndrome consisting of bilateral (rarely unilateral) hereditary retinoblastoma in association with an intracranial neuroblastic tumour arising usually in the pineal region, infrequently at the suprasellar or parasellar region. It can arise from either hereditary or sporadic forms of retinoblastoma. This article reports a child with uniocular retinoblastoma in association with suprasellar tumour and spinal metastases.
Trilateral retinoblastoma, Uniocular, Suprasellar tumor, Metastases
Retinoblastoma is the commonest primary ocular malignancy in children. Worldwide, the incidence is 1 case of retinoblastoma per 14,000 to 20,000 live births with some variation between countries [1]. On the other hand, trilateral retinoblastoma is a rare condition. Among 141 cases of retinoblastoma seen in Hospital Kuala Lumpur (the main referral centre for retinoblastoma in Malaysia) between August 2001 and December 2009, there were only two children with trilateral disease (1.4%) [2]. It is a disease characterized by a bilateral retinoblastoma (rarely unilateral) occurred in association with an intracranial neuroblastic lesion, which usually arise in the pineal region. Less frequently, the intracranial neuroblastic lesion may be detected at the suprasellar or parasellar region as well [1].
Trilateral retinoblastoma has poor prognosis for both ocular and survival outcomes. The prognosis is worse in developing countries where patients usually die of metastatic disease due to poor disease awareness and late presentation [3]. This article reports a child with uniocular retinoblastoma in association with suprasellar tumour and spinal metastases.
This is a 3-year-old boy who presented with 2 days history of headache, vomiting, lethargy and mild fever without neck rigidity. Retrospectively, the child's mother did notice white reflex of the left eye 1 year before the presentation but she did not seek any medical advice. There is no family history of retinoblastoma or other malignancy. On examination, there was left eye leucocoria and strabismus. There was no hypopyon, red eye or enlargement of the globe. Relative afferent pupil defect of the left eye was positive. Intraocular pressure was normal bilaterally. Visual acuity could not be assessed as the child was too ill. Fundus examination revealed a roundish whitish yellow mass about 2 cm in size, obscuring the temporal part of the disc which extended over the macular area with exudative retinal detachment further away from the macula. The nasal side of the disc was also blurry. Some blood streaks were noted within the lesion and blood vessels over it. There was no other satellite focus detected. The right eye was normal.
Computed Tomography (CT) scan showed a lobulated, hyperdensed intraocular fundus lesion in the left eye (Figure 1a) as well as a hyperdensed mass with specks of coarse calcification in the suprasellar intracranial mid line region (Figure 1b). Magnetic Resonance Imaging (MRI) showed left intraocular mass and retinal detachment without optic nerve or retro-orbital extension (Figure 2a). Brain and spine MRI also showed a highly cellular, bi-lobed suprasellar tumor with calcification (Figure 2b) which was complicated by moderate obstructive hydrocephalus, metastases at sphenoid, distal spinal cord, cauda equina and lumbosacral leptomeningeal regions.
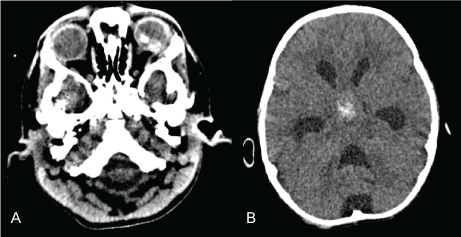 Figure 1: a) Orbit CT showing a lobulated, calcified, hyperdensed intraocular fundus lesion in the left eye; b) Brain CT shows a hyperdensed mass with specks of coarse calcifications in the suprasellar intracranial midline region.
View Figure 1
Figure 1: a) Orbit CT showing a lobulated, calcified, hyperdensed intraocular fundus lesion in the left eye; b) Brain CT shows a hyperdensed mass with specks of coarse calcifications in the suprasellar intracranial midline region.
View Figure 1
 Figure 2: a,b) Trilateral Retinoblastoma. Axial and sagittal MRI showing a mass in the suprasellar region with involvement of the left eye.
View Figure 2
Figure 2: a,b) Trilateral Retinoblastoma. Axial and sagittal MRI showing a mass in the suprasellar region with involvement of the left eye.
View Figure 2
Biopsy of the distal spinal cord tumor showed necrotic tumor tissue with foci of viable tumor clusters. The cells have round to oval nuclei, inconspicuous nucleoli and little cytoplasm. Immunohistochemistry showed reactivity patterns that are consistent with metastatic retinoblastoma, viz.: positive neuron-specific enolase and synaptophysin but negative glial fibrillary acidic protein.
The left eye enucleation was done under general anesthesia and histological examination showed retinoblastoma without optic nerve involvement. Histological features included viable tumor cells and necrotic tumor areas with calcification. There were also Flexner-Wintersteiner and Homer Wright rosettes.
The child also had right extraventricular drainage done to reduce the intracranial pressure. Subsequently the child was treated with a course of vincristine, carboplatin, and etoposide (JOE regime). However, the child died 2 months after the initial presentation due to progressive neurological disease.
Trilateral retinoblastoma is a rare condition, arising in about 3% of all the patients with retinoblastoma [4]. It manifests either as unilateral or bilateral intraocular retinoblastoma associated with an intracranial Primitive Neuroectodermal Tumour (PNET) either in the pineal, suprasellar or parasellar region. The association of intracranial neuroblastic lesion with unilateral intraocular retinoblastoma is less common than bilateral intraocular retinoblastoma with the incidence of 0.5% and 5%-15% respectively [1]. The intracranial lesions are considered to be independent primary retinoblastoma rather than metastatic spread as both retinal, and pineal or sellar tissue was originated from the same neural ectoderm [5].
Trilateral retinoblastoma can be diagnosed on the basis of eye examination, radiological imaging such as Computed Tomography (CT) scans and MRI, and then confirmed histologically. The diagnosis in our patient was challenging because he presented with concurrent retinoblastoma with intracranial and spinal lesion. Thus, the intracranial lesion needs to be differentiated between the primary tumor and metastatic lesion.
The diagnosis of trilateral retinoblastoma can be made and confirmed by CT scan and/or MRI. Cranial or orbital CT scans are able to detect intraocular calcifications and extent of the tumor [6]. MRI of the brain and orbits is the most sensitive means of evaluating for extraocular extension and it gives better delineation of the optic nerve and also the pineal area [6]. Our patient was diagnosed by CT and MRI, which is imperative to lower the possibility of misdiagnosis.
Intraocular retinoblastoma can extend to the brain by the optic nerve invasion or by spreading through the cerebrospinal fluid [7]. However, the suprasellar mass in our patient could not be considered as a metastasis from the retinal lesion because there was no involvement of the optic nerve or uveal tract as shown by ophthalmic examination and MRI as well as on histopathology of the left eye. It was reported that in trilateral retinoblastoma, the retinal tumor is usually small and there is often no optic nerve involvement [8]. Thus, both of these factors argue against cerebral metastasis.
Classically, cerebral metastasis represents multiple lesions with similar imaging characteristics, and the mid line location is uncharacteristic of the metastatic lesions [7].
The diagnosis of sellar trilateral retinoblastoma with spinal metastases was further confirmed by histopathology of enucleated left eye and spinal tumor biopsy. Histologically, retinoblastoma consists of cells with round, oval, or spindle-shaped nuclei that are hyperchromatic and surrounded by scanty cytoplasm, mitotic activity is usually high and Flexner-Wintersteiner rosettes are expressions of retinal differentiation [9].
The primary lesion is morphologically similar to intraocular retinoblastoma whereas metastatic retinoblastoma is characterized by undifferentiated small round cells without Flexner-Wintersteiner rosettes resembling neuroblastoma [9]. The histologic findings of spinal tumor in this patient resembled metastatic retinoblastoma.
Although the suprasellar trilateral retinoblastoma is diagnosed earlier than those with pineal trilateral retinoblastoma, their prognosis remains poor and the patients usually die of metastatic disease or postoperative complications within 10 months from the diagnosis [10]. According to several studies, the mean survival in patients with trilateral retinoblastoma varies from 1 to 28 months after diagnosis of the intracranial lesion [8,11].
In our case, the diagnosis of trilateral retinoblastoma remains a challenge because he presented with concurrent retinoblastoma with intracranial and spinal tumor. Trilateral retinoblastoma was diagnosed after the HPE of ocular tumor was confirmed as retinoblastoma and the biopsy of the distal spinal cord tumor are consistent with presence of retinoblastoma cells.