Gaucher disease (GD) type 1 is a lysosomal storage disorder associated with bone disease, hepatosplenomegaly, anemia and thrombocytopenia. Here we present a case series from 5 (3 females and 2 males) Portuguese individuals from a single institution with GD type 1 who were treated with substrate-reduction therapy (eliglustat, 84 mg once or twice daily) for 6-years. Four cases were switched from IV imiglucerase (28 U/kg q2 week [n = 1]) or 45 U/kg q2 week [n = 3]) and one was enzyme-replacement therapy naïve prior to eliglustat dosing. Two patients had 1304A > C (homozygous)/N435T mutations and the remainder had F148V/N409S mutations. GD Type 1 Severity Scoring System (GD-DS3) scores improved (n = 4) or remained stable (n = 1) over the 6-year follow-up. There were clear improvements in Gaucher Disease- Düsseldorf Gaucher Scores (GD- DGS) bone measures such as bone/joint pain and bone marrow burden (BMB) over follow up. Anatomical Düsseldorf Gaucher Scores improved (n = 4) or remained stable (n = 1). Mild thrombocytopenia in two cases at baseline both resolved. No splenomegaly or hepatomegaly was reported. The GD biomarkers chitotriosidase and ferritin decreased in all patients over follow-up. These data confirm long-term treatment trends in GD type 1 patients treated with eliglustat. Eliglustat achieved recommended treatment goals such as improvement or maintenance of GD scores, maintenance of visceral measures such as spleen and liver size, the reduction of hematological measures such as thrombocytopenia/anemia and improvements in GD biomarkers over 6-years of follow up.
Eliglustat, Enzyme replacement therapy, Gaucher disease, Glucocerebrosidase, Imiglucerase, Lysosomal storage disorders
Gaucher disease (GD) is a lysosomal storage disorder reflecting mutations in the glucocerebrosidase (GCase) gene (GBA1), which codes for the enzyme that breaks down glucosylceramide (GlcCer). This causes GlcCer-laden Gaucher cells to build up in the spleen, liver and bone marrow. How ClcCer and glucosyl sphingosine (LysoGL-1) produced by extralysosomal GB2 may contribute to GD type 1 pathophysiology and variable phenotype is not completely understood [1]. GD type 1 is the most common form, accounting for approximately 90% of cases in Europe and the USA, and is associated with bone disease, hepatosplenomegaly, anemia and thrombocytopenia, lung disease, but no primary central nervous system disease [2,3].
The clinical presentation of GD type 1 is variable ranging from asymptomatic to early-onset forms that present in childhood, however, most individuals experience symptoms before the age of 20. While over 300 mutations in the GBA1 gene have been described, genotype-phenotype correlations are poorly characterized, particularly in relation to disease severity and progression [2]. There is evidence of bone disease in > 70% of individuals with GD type 1. These include focal lytic or sclerotic lesions and osteonecrosis, often associated with acute or chronic pain, which can lead to joint collapse and arthritis [3]. Fatigue affects around half of individuals and can interfere with school and working life. Splenomegaly is present in approximately 90% of cases and in some cases spleen size may directly cause abdominal pain [2]. Hepatomegaly affects 60-80% of individuals with GD type 1 but the development of fibrosis, cirrhosis or liver failure are rare [2,3].
GD is rare in the general population affecting 1 in 40,000 to 60,000 live births. The incidence is higher in populations such as those with Ashkenazi Jewish descent, in which GD occurs in approximately 1 in 800 births due to a founder effect [2,3]. Another population with above normal incidence of GD has been reported in Portugal, which is also thought to relate to a common ancestral founder [4].
There are two main forms of treatment for GD, enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) which acts by to reducing the production of GlcCer [2,3]. Here we present a case series from 5 Portuguese individuals with GD type 1 who have been treated with the oral SRT eliglustat for 6-years.
This is a case series of 5 adult patients, 3 females and 2 males, with GD type 1 from a single institution who were treated with oral eliglustat, with 6-years of follow-up data. All patients provided written informed consent for their case details to be presented. The patients were born between 1953 and 1966 and were diagnosed between 26 and 57 years of age. Four patients had previously been treated with ERT for between 4 and 15 years and 1 patient was ERT-treatment naïve (Case 1).
Two patients had 1304A > C(homozygous)/N435T (traditional nomenclature (1304A > C(homozygous)/N396T) mutations which is generally associated with a mild phenotype and occurs in 5.6% of Portuguese Gaucher population. The remaining three patients had the F148V/N409S (traditional nomenclature F109V/N370S) mutations. F109V is a rare mutation associated with 7% residual acid α-glucosidase activity and a mild phenotype. Only one patient had undergone total splenectomy and cholecystectomy eleven years prior to the initiation of ERT and twenty-one years prior to oral SRT eliglustat (Case 5).
Patient 1 had a number of co-morbidities including type 2 diabetes that required insulin treatment, diabetic nephropathy with mild chronic renal impairment, hypertension, hyperuricemia, dyslipidemia, uterine myoma, carpal tunnel syndrome and fibroadenoma of the breast. In total, 12 medications were required to treat comorbidities. Given that eliglustat is metabolized by CYP2D6 and to a lesser extent by CYP3A4 and that it inhibits P-gp and CYP2D in vitro, care should be taken with concomitant administration of CYP2D6 or CYP3A4 inhibitors or substrates for P-gp. Despite this, eliglustat was able to be prescribed in this patient.
Patient 2 had a cardiac condition (incomplete left bundle branch block) prior to initiating eliglustat that progressed to complete left bundle branch block following eliglustat treatment. While European and US recommendations advise against the use of eliglustat in patients with cardiac comorbidities including heart block, it was decided in close consultation with a cardiologist that the patient would still benefit from eliglustat treatment. Patient 2 also reported depression post-eliglustat treatment. Patient 3 had type 2 diabetes adequately treated with oral drugs, without short or long term complications.
Of the five cases, four were switched from ERT imiglucerase (28 U/kg q2wk [n = 1]) or 45 U/kg q2wk [n = 3]) (Figure 1). The final patient was ERT naïve prior to eliglustat. A key reason for the choice of eliglustat was that it is taken orally at home, whereas imiglucerase is administered intravenously and in Portugal requires a hospital visit every 2 weeks. All 5 patients started eliglustat in July or August 2011. Four patients were extensive CYP450 2D6 metabolizers (EMs) and one patient (F148V/N409S mutation) was an intermediate metabolizer (IM), but had a clinical phenotype of a poor metabolizer based on peak plasma levels of eliglustat (Case 5). The recommended dose of 84 mg eliglustat twice daily in CYP2D6 EMs, and 84 mg eliglustat once daily in CYP2D6 poor metabolizers, was followed in this case series. Eliglustat has only been in commercial use since 2014. Adverse events reported from eliglustat clinical trials were mild or moderate, transient, and occurred only once per patient [5].
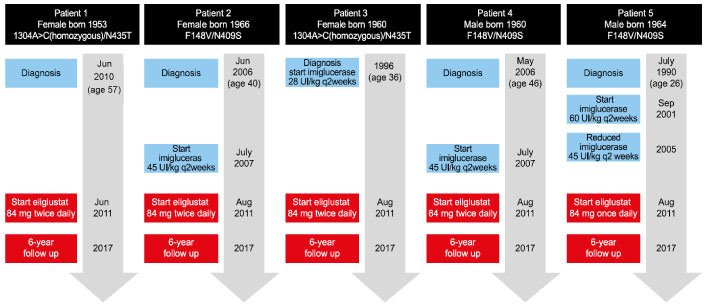 Figure 1: Case timelines.
View Figure 1
Figure 1: Case timelines.
View Figure 1
Clinical parameters measured at treatment initiation and after 6-years of follow up included bone markers (number of lytic lesions/avascular osteonecrosis (AVN)/fractures; episodes of joint pain within past 30 days; bone marrow crisis past 12 months, Düsseldorf score; bone domain score, BMD lumbar Z-score (three Radiologists reviewed all the images [MRI bone and visceral] individually, discussed and provided a consensus opinion), hematological markers, visceral measurements, GD-DS3 [6] and biomarkers including chitotriosidase (CHITO), [7] and ferritin. MRI provide semiquantitative assessment of marrow infiltration and is the gold standard for monitoring bone involvement. In this case series, all radiologic imaging were assessed by the same equipment (dual-energy X-ray absorptiometry [DXA]- GE-Healthcare Lunar® and a MRI - Siemens®). Osteonecrosis was defined on the basis of conclusive evidence on MRI or plain radiologic imaging [8].
At baseline, four of 5 patients had DS3 scores indicative of moderate GD (DS3 3.0 to 4.3) and the remaining patient had a DS3 score of 2.7, indicating mild disease (Table 1). Cases 2, 4 and 5 had bone complications including lytic lesions, AVN, or fractures at baseline. Over the 6 years of follow up GD-DS3 scores either remained stable or decreased, despite eliglustat treatment interruption in two cases due to hospitalization for emergency appendectomy (Case 2) or total hip prosthesis (Case 4), both cases GD-DS3 severity score decreased from moderate to mild. In Case 1, ERT naïve prior to eliglustat patient, GD-DS3 severity score classification decreased from moderate to mild.
Table 1: Gaucher Disease Type 1 Severity Scoring System (GD-DS3) before and after 6-years of eliglustat treatment. View Table 1
There were clear improvements in the bone components of GD-DS3 such as bone/joint pain and bone marrow infiltration during follow up (Figure 2 and Figure 3). At eliglustat initiation, 3 patients reported moderate or severe bone pain in the previous 30 days, however, at the 6-year follow up these patients reported mild or no/very mild bone or joint pain. Bone marrow infiltration (bone marrow burden [BMB]) was also reduced from marked/severe to moderate in Cases 1 and 2, remained moderate in Case 3 and remained marked/severe in Cases 4 and 5. Prior to eliglustat initiation, Patient 1 had experienced two bone crises in the previous 12 months, whereas no bone crises were reported in any patient following eliglustat initiation.
 Figure 2: Coronal T1 - weighted images MR images of tibia of a female Gaucher patient. A) Tibia before switch from imiglucerase to eliglustat. The images obtained detected an abnormal focal changes marrow bone - focal low signal intensity in both diaphysis, sparing the epiphysis and metaphysic; B) After 6y of eliglustat therapy, images of the same patient noted there was increased bone marrow signal in the diaphysis, compared with the baseline, with no lesions detected in diaphysis.
View Figure 2
Figure 2: Coronal T1 - weighted images MR images of tibia of a female Gaucher patient. A) Tibia before switch from imiglucerase to eliglustat. The images obtained detected an abnormal focal changes marrow bone - focal low signal intensity in both diaphysis, sparing the epiphysis and metaphysic; B) After 6y of eliglustat therapy, images of the same patient noted there was increased bone marrow signal in the diaphysis, compared with the baseline, with no lesions detected in diaphysis.
View Figure 2
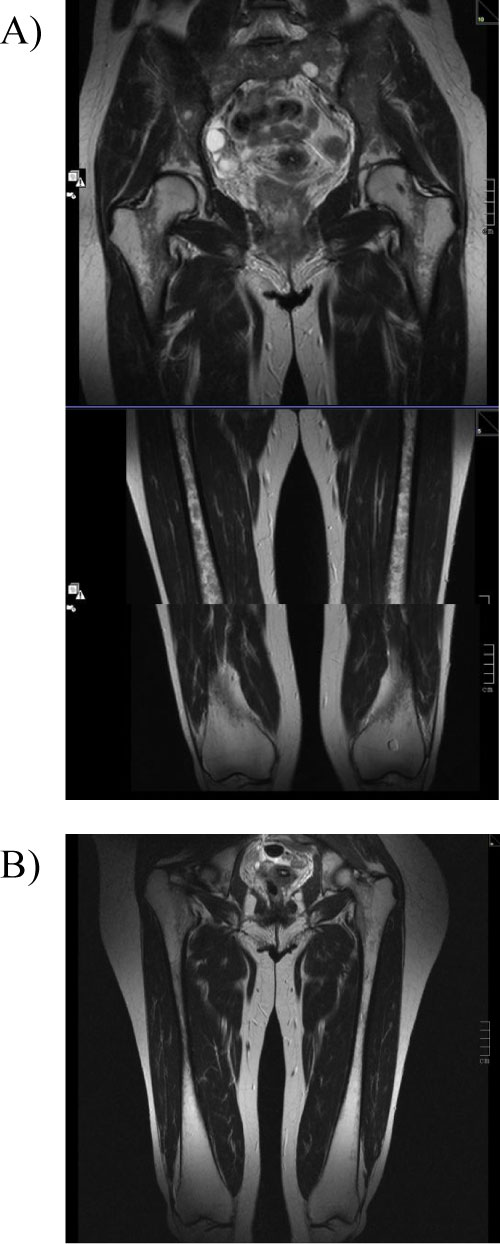 Figure 3: Coronal T2 - weighted images MR images of bilateral femur.
Figure 3: Coronal T2 - weighted images MR images of bilateral femur.
A) Femur before switch from imiglucerase to eliglustat. Images clearly show diffuse homogeneous hypointensity in diaphysis. BMB score 7 for the femur. B) After 6y of eliglustat therapy. Images After 6y of eliglustat therapy, images indicate BMB score of 4 for the femur. Proximal epiphysis slightly hypointense on T2, indicating a marrow response following medication with eliglustat. Initial Düsseldorf score of 8B improved to 3B after 6 years of treatment (increased bone marrow signal in the tibias [not shown in the present illustration] while both metadiaphysis of the femur are still affected).
View Figure 3
The Düsseldorf Gaucher Score (DGS) is a semi-quantitative Magnetic Resonance Imaging (MRI) based system [9], which allows scoring of Gaucher cell bone-marrow infiltration of lower extremities and for differentiating between A-patterns (homogeneous) and B-patterns (non-homogenous) of infiltration [10]. The DGS score is based on the number of femoral segments with infiltration, and the highest numbered site of involvement is taken as a measure of disease severity. Anatomical DGS scores were reduced in four cases and remained stable in one case (Case 5), whereas morphological DGS scores were B (non-homogeneous) in all cases at both initiation and 6-year follow-up (Table 1). A 4-year follow up already showed DGS scores were reduced in three cases and stable in two cases (Case 4 and 5).
Thrombocytopenia normalized in the two cases who were mildly thrombocytopenic at eliglustat initiation. There was no thrombocytopenia in the remaining 3 cases (Table 1).
No bleeds or anemia cases were reported prior to, or post, eliglustat treatment (Table 1).
No cases of splenomegaly or hepatomegaly were reported prior to, or post, eliglustat treatment (Table 1). At initiation, in the 4 cases in which spleen size was reported (Cases 1-4) size varied between 1.9 and 3.2 times of normal, which was not categorized as splenomegaly. Similarly, liver size at baseline was between 0.7 and 1.0 times of normal.
Chitotriosidase (CHITO): Gaucher cells produce CHITO, so it is useful for monitoring treatment efficacy and may have prognostic value [2]. CHITO levels can vary considerably among patients so it is not useful for between-patient comparisons. In three patients (1, 4 and 5) CHITO levels fell over 80%, while in patients 2 and 3 levels fell 46% and 68%, respectively, at 6-years following eliglustat treatment (Table 1). CCL18/PARC concentration, is not performed in Portugal. And, baseline, serum biomarker Glucosyl sphingosine data was not available.
Ferritin: Higher than normal levels of ferritin can be observed in individuals with GD [2]. Eliglustat treatment was associated with ferritin reductions of between 57% and 94%.
This case series of 5 patients provides long-term follow up over 6-years following the initiation of eliglustat. The strengths of these data include the comprehensive data on clinical characteristics and outcomes that were collected, and the long-term 6-year follow up.
Recent expert consensus publications recommended that treatment should reduce bone marrow involvement measured by a system such as the BMB or DGS in patients without severe irreversible bone disease at baseline [11]. This was achieved in this case series with either improved (n = 4) or stable (n = 1, patient 5) DGS scores being recorded over 6-years. By MRI analysis, GD-DS3 scores indicated improvement in 4 of the 5 patients of their anatomical scores while the same individual who had a stable DGS score (patient 5) had a stable GD-DS3 score. Similarly, BMB scores also improved (n = 2) or remained stable (n = 3) over the 6-year follow up. These results confirm in daily clinical practice the results reported in clinical trials, which have shown improvements in bone mineral density and bone marrow infiltration following eliglustat treatment [12-14].
In this case series, 3 patients before switching from ERT to SRT - eliglustat, already presented bone infarcts (Case 2, 4 and 5). After 6 year follow-up none developed new bone manifestation.
Splenectomy is an important risk factor for osteonecrosis and splenectomized GD patients revealed higher BMB scores than non-splenectomized GD patients [15]. In this case series, the splenectomized patient remainded stable DGS score and BMB score (slight improvement femur infiltration) and showed improvement in bone mineral density, after 6-years on oral SRT eliglustat.
Clinical trials have also reported sustained improvements in visceral measures such as liver and spleen volumes with eliglustat treatment [12-14]. Since there was no evidence of splenomegaly or hepatomegaly at baseline in this group of patients, it can be concluded from this case series that there was no worsening of these measures over the 6-year follow up. Consensus guidelines recommend that ERT should be associated with a reduction in spleen volumes of < 2 to 8 times normal size. This recommendation was achieved in patients 1-4 who had spleen measurements between 1.9 and 3.2 times of normal at eliglustat initiation and no indication of splenomegaly at 6-year follow up. Similarly, liver volumes were between 0.7- and 1-times normal size at eliglustat initiation in 4 of 5 patients (the final patient had no hepatomegaly but liver size was not documented), and none of the five patients had hepatomegaly at 6-year follow up, which is consistent with consensus guidelines. The long-term aims of GD treatment should also be to prevent or improve pulmonary disease, such as pulmonary hypertension and hepatopulmonary syndrome [11] and no pulmonary disease was observed either at baseline or the 6-year follow up in any patient.
GD also includes a hematologic burden of thrombocytopenia, anemia and bleeding. Consensus guidelines recommend eliminating anemia-related symptoms and the need for blood transfusion, however, anemia was not observed in this group of patients at baseline or during follow-up. Guidelines also recommend that platelet counts be normalized [11], and this goal was achieved in the 2 patients who had thrombocytopenia at baseline. No bleeds were observed at baseline or during follow up.
The improvements in GD biomarkers observed over the 6-year follow up were consistent with results reported in eliglustat clinical trials. For example, the reduction in CHITO levels of between 46% and 83% is comparable to the 63% reduction over 4 years reported in the phase 3 ENCORE trial [13]. Ferritin levels were also substantially reduced by between 57% and 94% over the 6-year follow up.
Daily quality-of-life (QoL) is also an important goal for GD therapy with consensus guidelines recommending that measurement be performed with a validated QoL instrument, that fatigue is measured using a validated scoring system, that individuals can maintain participation in school or work activities and have a normal life expectancy [11]. While the case analysis included measures that reflect QoL such as bone/joint pain and bone crises, a limitation of this case analysis is that validated QoL measures were not included.
Patients had mild and transient adverse events to oral eliglustat, arthralgia metacarpo-phalangeal and proximal interphalangeal joints (Case 4); abdominal distension (Case 5). Only patient 1 persists with mild dry skin (Case 1).
In conclusion, these data provide useful information on long term treatment trends in type 1 patients treated with oral eliglustat. Eliglustat treatment achieved recommended treatment goals such as improvement or maintenance of GD scores, maintenance of visceral measures, the reduction of hematological measures and improvements in GD biomarkers over 6-years of follow up.
Case-study data from 6-years of oral eliglustat treatment in 5 patients with Gaucher disease type 1 indicates long-term improvements in disease scores, bone measures and biomarkers.
Cristina Fraga treated all 5 patients and collected data at baseline and follow up. Radiologists Sónia Medeiros, Sara Serpa, David Silva assisted with radiological imaging, reviewed all the images (MRI bone and visceral) analyzed images individually and reached a consensus opinion in collaboration with the other Radiologists. All authors were involved with the preparation of the manuscript, critical review of the manuscript at all stages and approved the final version prior to submission.
Cristina Fraga has received honoraria for speaker services, participation on advisory boards and travel reimbursement from Sanofi Genzyme. Sónia Medeiros, Sara Serpa, David Silva declare no conflicts of interest.
Editorial assistance was provided by Michael Lappin of GK Pharma Comm Ltd.
Ethical approval was not required for the case studies.
Patients provided written informed consent for their case details to be presented.