Purpose: Paraganglioma is a neural crest-derived endocrine cells or organs located generally at the extra-adrenal glands, head, necks, or abdomen. This research case study brings a detailed information of paraganaglioma occurred for a patient in Aseer region.
The sample: 36-year-old patient who experienced no major symptoms such as hypertension, palpitation, vomiting, bleeding, or headache.
The tools: for primary diagnosis ultrasonography and Computed Tomography (CT) were used to identify a heterogenous mass. The Magnetic Resonance Imaging (MRI) was used to further validate the primary findings and histo-pathologic test after getting out the mass.
Conclusions: The study confirms that the patient had a paraganglioma with vascular connected tissues inside the mass free from tumor invasion and its surgical margins are free from tumor cells. The study supports the fact that such paraganglioma with patient is free from any symptoms is hereditary.
Paragangliomas, Extra-adrenal gland, Tumor invasion
It is commonly known that paragangliomas are rare tumors and most of the cases have approved to be benign and some are malignant. After examination of the clinical characteristics, location, treatment, and outcome of 236 patients it was found that 141 females, (60%) with 297 benign paragangliomas evaluated at the Mayo Clinic during 1978-1998 [1]. It was found that a clinical follow-up of 98 cases where 64 tumors were clinically benign, and 34 were malignant as evidenced by regional or distant metastases and/or extensive local invasion where the thirty-two of the 34 malignant tumors (94%) were functionally active [2]. Studies in this regard have been done to show the emergence of paragangliomas where they can be found in the neck [3], head [4,5], the extra-adrenal glands or abdomen [6-8]. Many tumors of such type, paragangliomas and Pheochromocytomas are diagnosed to be of hereditary tumors type [9-11]. In a genetic study of 314 patients with Pheochromocytomas and paragangliomas, it was identified that 86 patients (27.4%) with a hereditary tumor [12]. Similarly another study showed that Pheochromocytomas and paragangliomas carry the highest degree of heritability (around 40%) of all human tumours and thus represent relevant models for the identification of driver mutations in cancer [11]. It also explored that Thirty per cent of the paragangliomas and pheochromocytomas reported are hereditary [13]. They are to great extent associated with the autonomous nerve system of the body.
We report a rare case of a patient with benign retroperitoneal Paraganglioma in the external-adrenal gland was seen in Asir Central Hospital. The medical record of the patient was reviewed and the follow-up data was obtained. 36-year-old patient experienced no major symptoms such as hypertension, palpitation, vomiting, bleeding, or headache. At the beginning, the physicians thought that the patient was having Pheochromocytomas and ordered for some tests including (Catecholamine in Plasma): 1) Epinephrine; 2) Norepinephrine; 3) Dopamine. The test was to avoid any crisis during the surgery as the tumor was close to the external adrenal gland. The results showed that the patient was not having Pheochromocytomas as the epinephrine was < 40 pg/ml, Nor-epinephrine 708 pg/ml, and dopamine < 20 pg/ml.
Firstly, Ultrasonography showed round retroperitoneal mass measuring approximately 7.8 × 6.6 cm in left region of abdomen anterior to left kidney. Then a plain Computed Tomography (CT) and contrast-enhanced axial CT were suggested.
Figure 1 of a) Plain Computed-Computed Tomography (CT) and B) Contrast-enhanced axial CT scan of the upper abdomen showed a homogenous mass measuring 7.2 × 5.7, seen in the left lumbar aspect of the abdomen. The follow up Figure 2 MRI showed retroperitoneal soft tissue mass close to the origin of the IMA (Expected location of ZUckerkamdl's body) at the left para aortic region measuring 95 × 75 mm in its maximum dimensions. It explored internal areas of breakdown. The peripheral lesion shows T1 iso-hypointensity and T2 hyperintensity with intense vascular blush and multiple dilated retroaortic vascularity within and outside the lesion. The described features are those of retroperitoneal highly vascular soft tissue neoplastic lesion could be a paraganglioma (extra-adernal pheochromocytoma). And the surgical excision was the only treatment choice to be done in this stage and the surgical procedure was decided by the medicine physicians soon and a large homogeneous mass is clearly shown during the intraoperative process as in Figure 3.
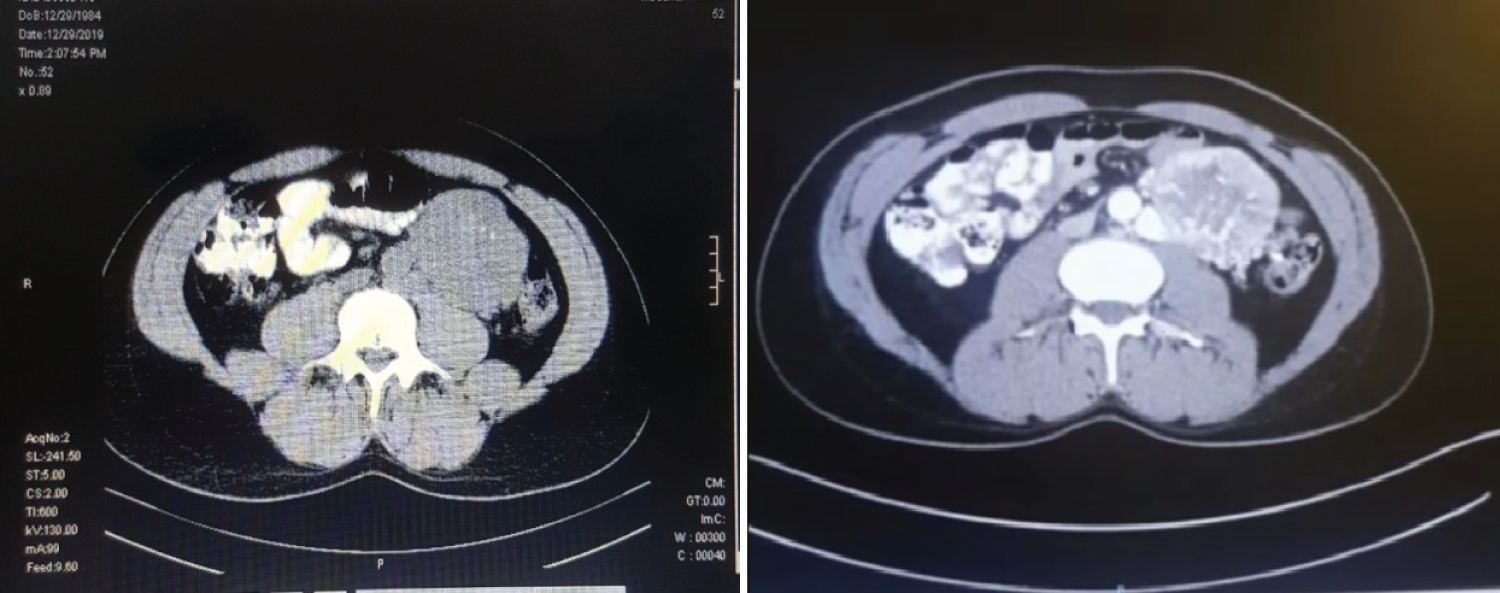 Figure 1: A) A plain Computed-Computed Tomography (CT) and B) contrast-enhanced axial CT scan of the upper abdomen showed a homogenous mass measuring 7.2 × 5.7, seen in the left lumbar aspect of the abdomen.
View Figure 1
Figure 1: A) A plain Computed-Computed Tomography (CT) and B) contrast-enhanced axial CT scan of the upper abdomen showed a homogenous mass measuring 7.2 × 5.7, seen in the left lumbar aspect of the abdomen.
View Figure 1
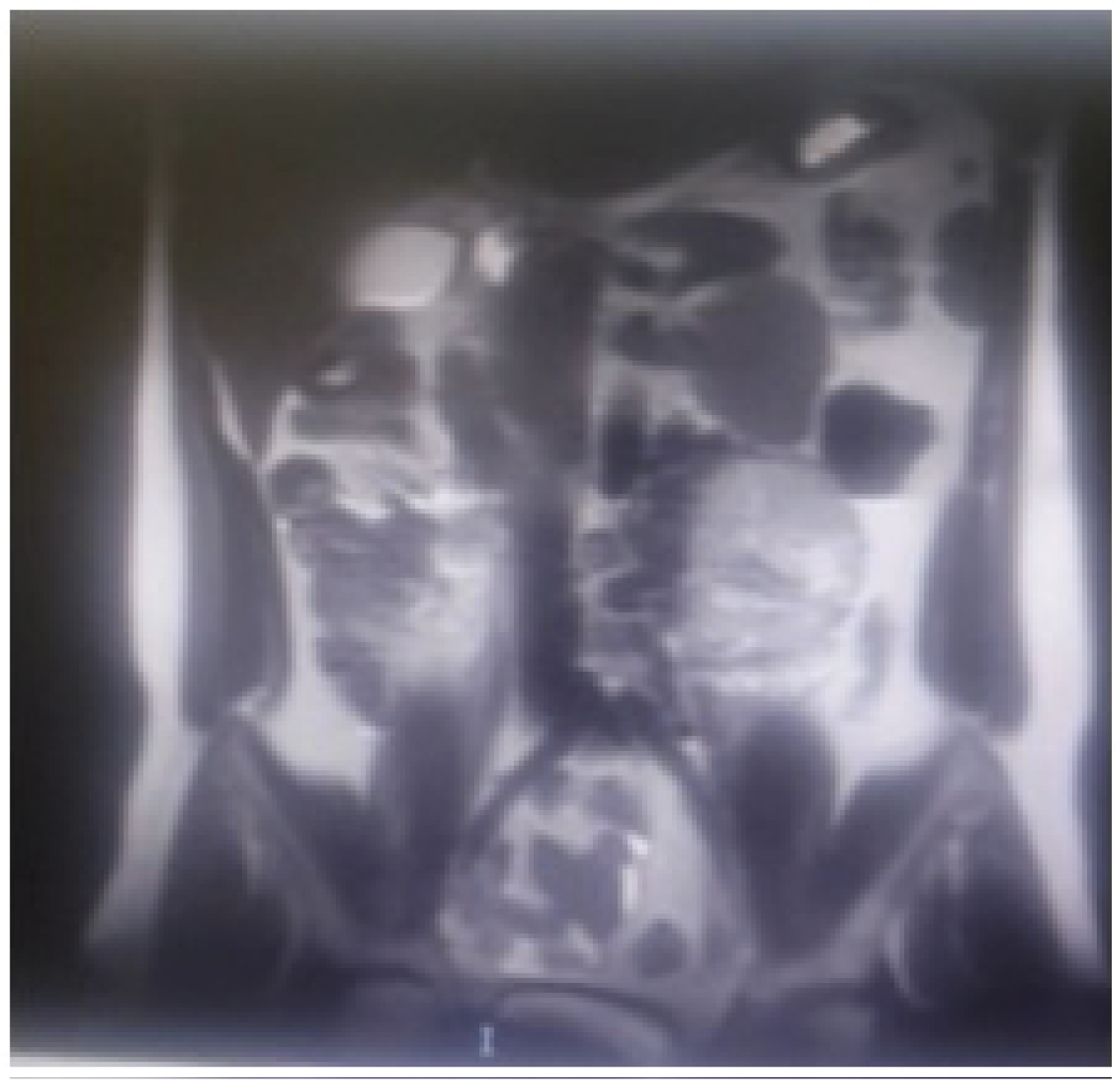 Figure 2: MRI showed a retroperitoneal soft tissue mass near the origin of the IMA (Expected location of ZUckerkamdl's body) at the left para aortic region measuring 95 × 75 mm.
View Figure 2
Figure 2: MRI showed a retroperitoneal soft tissue mass near the origin of the IMA (Expected location of ZUckerkamdl's body) at the left para aortic region measuring 95 × 75 mm.
View Figure 2
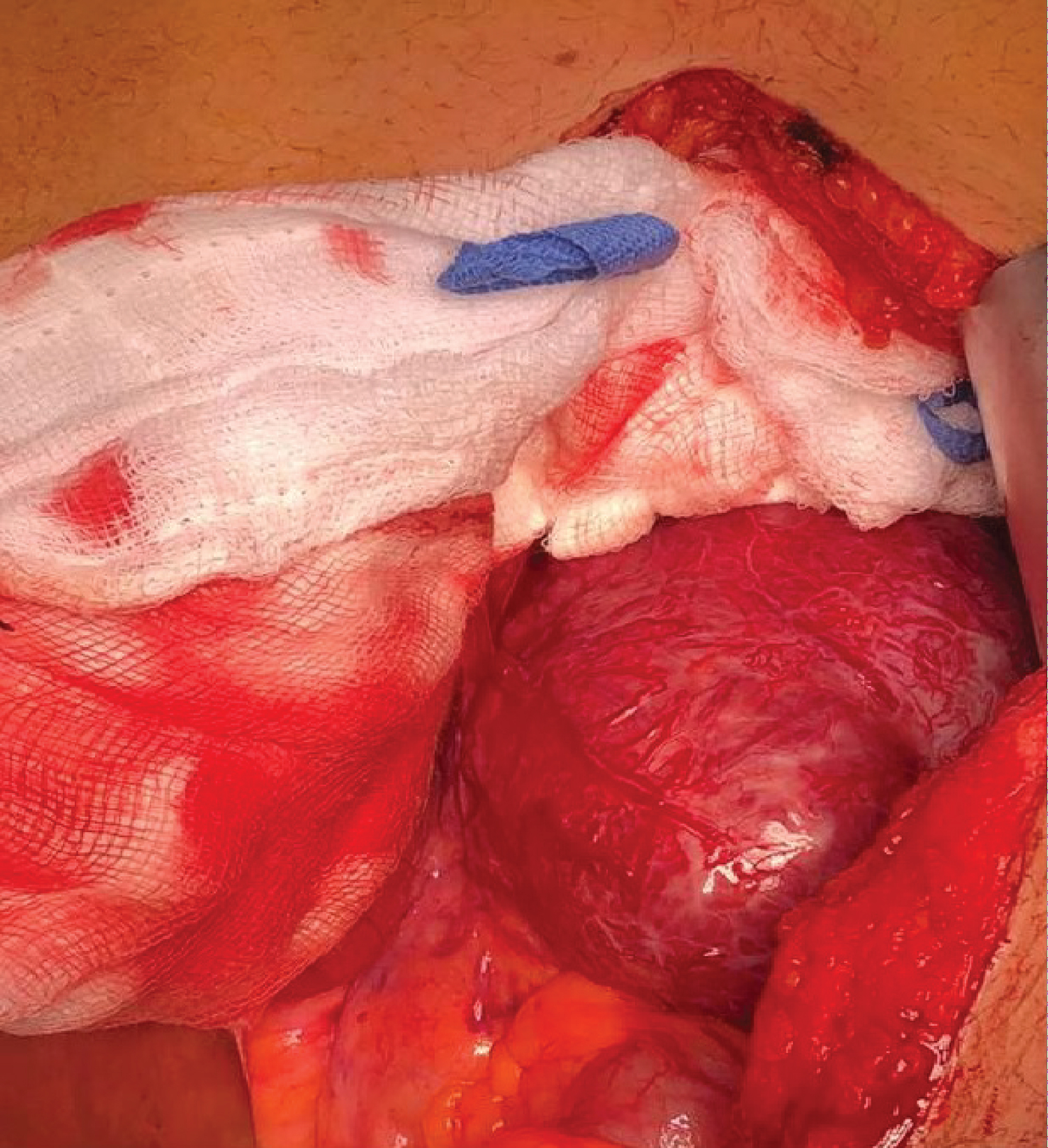 Figure 3: Intraoperative figure showing the mass in the left part of abdomen as one part with egg shell calcification.
View Figure 3
Figure 3: Intraoperative figure showing the mass in the left part of abdomen as one part with egg shell calcification.
View Figure 3
The histo-pathologic test supported the fact that the tumor, retroperitoneal mass, was paraganglioma measuring 10 × 7.5 × 5 cm. The mass was with mainly cellular nests and trabecular pattern of cells within prominent vascular network focally obscured by anastomosing bands of cells. The mass is free of tumor invasion.
After a month of post-treatment, the patient is able to eat solids easily and after the clinical process continued no external mass in the previous location, at the left para aortic region with three-months of follow up. This paraganglioma in our case can caused as hereditary. The opposed many studies which support the view that it can be due to excess secretion of catecholamine.
To sum up, though there limited ways to detect paragangliomas at the preoperative stage surgery stays the best treatment approach. This case study brings to the literature that paraganliomas are hereditary. Due to the silent growths of paraganglioma and its rarity in the literature, this case report could help as a hint of suspected paragangliomas. Follow-up is highly recommended to check the reoccurrence of the similar mass. Further collaborative studies with larger sample are recommended to test the nature of paragangliomas in the same region and investigating the additional causes of such tumors.